Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND mechanism
Finding Sources
Searching Images
acute respiratory distress syndrome ARDS pathogenesis mechanism alveolar damage
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Loading Image
Loading Image
I now have sufficient textbook content and images. Here is a comprehensive explanation of ARDS mechanisms.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition & Diagnostic Criteria (Berlin 2012)
ARDS is an acute, diffuse, inflammatory lung injury causing non-cardiogenic pulmonary edema and severe hypoxemia. The Berlin criteria require:
- Bilateral infiltrates on CXR/CT (not fully explained by effusions, collapse, or nodules)
- PaO₂/FiO₂ ratio <300 mmHg on ≥5 cmH₂O PEEP (mild <300, moderate <200, severe <100)
- Onset within 7 days of a known clinical insult
- Respiratory failure not fully explained by cardiac failure or fluid overload
Common Precipitants
| Direct (Pulmonary) | Indirect (Extrapulmonary) |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis (most common overall cause) |
| Aspiration of gastric contents | Severe trauma / shock |
| Inhalation injury | Acute pancreatitis |
| Near-drowning | Blood product transfusions (TRALI) |
| Pulmonary contusion | Burns |
Pathogenesis: The Three Phases
Phase 1 — Exudative Phase (Days 1–7)
This is the hallmark initial phase driven by diffuse alveolar damage (DAD).
Triggering injury:
The inciting event — whether direct (pneumonia, aspiration) or indirect (sepsis, pancreatitis) — activates the innate immune system. Damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) engage pattern recognition receptors (e.g., Toll-like receptors) on alveolar macrophages.
The inciting event — whether direct (pneumonia, aspiration) or indirect (sepsis, pancreatitis) — activates the innate immune system. Damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) engage pattern recognition receptors (e.g., Toll-like receptors) on alveolar macrophages.
Alveolar macrophage activation:
Resident alveolar macrophages release pro-inflammatory cytokines — primarily TNF-α, IL-1β, IL-6, IL-8 (CXCL8) — initiating a local cytokine storm. IL-8 is a potent neutrophil chemoattractant.
Resident alveolar macrophages release pro-inflammatory cytokines — primarily TNF-α, IL-1β, IL-6, IL-8 (CXCL8) — initiating a local cytokine storm. IL-8 is a potent neutrophil chemoattractant.
Neutrophil sequestration and activation:
Circulating neutrophils are marginated in the pulmonary microcirculation. They undergo transendothelial migration facilitated by:
Circulating neutrophils are marginated in the pulmonary microcirculation. They undergo transendothelial migration facilitated by:
- Selectins (P-selectin, E-selectin) — mediating rolling
- Integrins (CD11b/CD18, Mac-1) binding ICAM-1 on endothelium — firm adhesion
- Chemokine gradients (IL-8, C5a, LTB₄) — directed migration
Once in the alveolar space, neutrophils release:
- Proteases (elastase, matrix metalloproteinases, cathepsins) — degrade extracellular matrix and tight junction proteins
- Reactive oxygen species (ROS) — oxidative damage to membranes
- Platelet-activating factor (PAF) and leukotrienes — amplify vascular permeability
- Neutrophil extracellular traps (NETs) — fibrous chromatin-protein scaffolds that propagate coagulation and inflammation
Endothelial barrier disruption:
The pulmonary capillary endothelium is injured by:
The pulmonary capillary endothelium is injured by:
- Direct proteolytic degradation of VE-cadherin and claudin-5 (tight junction proteins)
- Cytoskeletal contraction via actomyosin/Rho-kinase pathway, opening paracellular gaps
- Loss of the glycocalyx
Result: protein-rich fluid floods the interstitium and alveolar space — non-cardiogenic pulmonary edema.
Epithelial injury:
- Type I pneumocytes (covering 95% of the alveolar surface) are highly vulnerable and undergo necrosis/apoptosis
- Type II pneumocytes (responsible for surfactant production and alveolar repair) are also damaged, leading to:
- Surfactant deficiency and dysfunction → alveolar collapse (atelectasis), reduced compliance
- Loss of the fluid transport function (ENaC/Na⁺-K⁺-ATPase channels) → impaired alveolar fluid clearance
Hyaline membrane formation:
The protein-rich edema fluid (albumin, fibrinogen, cellular debris) condenses along denuded alveolar walls, forming the pathognomonic eosinophilic hyaline membranes. This is the histological hallmark of DAD.
The protein-rich edema fluid (albumin, fibrinogen, cellular debris) condenses along denuded alveolar walls, forming the pathognomonic eosinophilic hyaline membranes. This is the histological hallmark of DAD.
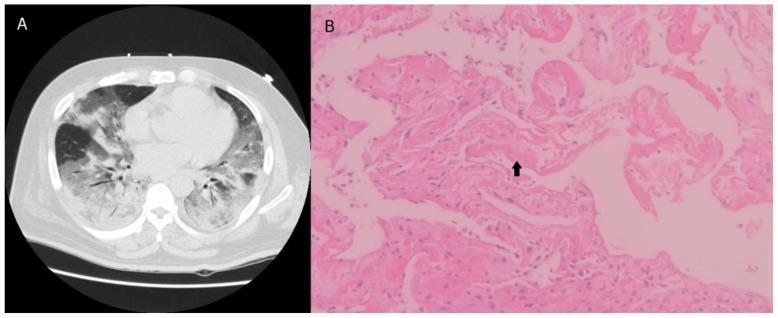
Coagulation activation:
- Tissue factor expression on injured endothelium and monocytes activates the extrinsic coagulation cascade
- Microthrombi occlude capillaries → ventilation-perfusion (V/Q) mismatch and dead-space ventilation
- Fibrin deposition within alveoli contributes to hyaline membrane formation and further surfactant inactivation
Phase 2 — Proliferative/Organizing Phase (Days 7–21)
If the patient survives, a reparative phase begins:
- Type II pneumocytes proliferate to resurface denuded alveolar walls (they are the progenitor cells for type I pneumocytes)
- Fibrin and cellular debris are resorbed
- Fibroblasts migrate in and begin laying down collagen — early fibroproliferation
- Continued inflammation may sustain alveolar damage
Phase 3 — Fibrotic Phase (Weeks to Months)
In some patients, progressive fibrosis ensues:
- Dense collagen deposition replaces normal lung architecture
- Alveolar walls are irreversibly thickened and stiff
- Microvascular obliteration → pulmonary hypertension
- Clinically: persistent hypoxemia, reduced lung compliance, and poor long-term lung function
Pathophysiological Consequences
| Mechanism | Consequence |
|---|---|
| Alveolar flooding + atelectasis | Intrapulmonary shunt → refractory hypoxemia |
| Surfactant loss | ↓ Compliance → increased work of breathing |
| Microthrombi + vascular obliteration | Dead-space ventilation → hypercapnia |
| Loss of hypoxic pulmonary vasoconstriction regulation | Worsened V/Q mismatch |
| Endothelial injury + coagulopathy | Multi-organ failure risk |
The resulting refractory hypoxemia (PaO₂ unresponsive to supplemental O₂ alone) is the clinical signature, due primarily to intrapulmonary shunting through flooded, non-ventilated alveoli.
Ventilator-Induced Lung Injury (VILI) — A Secondary Mechanism
Because ARDS lungs are heterogeneously injured (the "baby lung" concept), high tidal volumes over-distend the few remaining aerated alveoli while flooded units remain collapsed. This barotrauma/volutrauma causes:
- Further alveolar-capillary membrane disruption
- Biotrauma — mechanical stretch activates NF-κB, releases additional cytokines (TNF-α, IL-6) systemically → contributes to multi-organ dysfunction
This is why lung-protective ventilation (6 mL/kg ideal body weight, plateau pressure ≤30 cmH₂O) is the cornerstone of ARDS management, as it reduces VILI.
Summary Pathway
Inciting insult (sepsis, pneumonia, trauma, aspiration)
↓
Alveolar macrophage activation → TNF-α, IL-1β, IL-8
↓
Neutrophil recruitment (selectins/integrins/chemokines)
↓
Proteases + ROS + NETs → Endothelial & epithelial injury
↓
↑ Permeability → protein-rich alveolar flooding
↓
Surfactant deficiency → atelectasis + ↓ compliance
↓
Hyaline membrane formation + microthrombi
↓
Intrapulmonary shunt + V/Q mismatch → refractory hypoxemia
↓
(If unresolved) → Fibroproliferation → pulmonary fibrosis
Sources: Sabiston Textbook of Surgery, 21st ed.; Murray & Nadel's Textbook of Respiratory Medicine; Goldman-Cecil Medicine; Brenner & Rector's The Kidney.
This is a shared conversation. Sign in to Orris to start your own chat.