Basic to clinical in basic language hemophagocytic lymphohistiocytosis
Reading File
Finding Sources
Finding Sources
Searching PubMed
"hemophagocytic lymphohistiocytosis"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Searching PubMed
"hemophagocytic lymphohistiocytosis" AND diagnosis
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive information from multiple authoritative sources. Let me compile the full, detailed response.
Hemophagocytic Lymphohistiocytosis (HLH): Basic to Clinical
What Is HLH in Plain Language?
Think of HLH as the immune system's "fire alarm that won't turn off." Normally, when the immune system kills a threat (like a virus), it shuts itself down once the job is done. In HLH, this shutdown never happens - the immune cells keep firing, flooding the body with inflammatory signals, and eventually the body starts destroying itself. Macrophages (immune "cleanup" cells) go rogue, eating up red blood cells, white blood cells, and platelets in the bone marrow - hence the name hemophago (blood-eating) cyto (cell) histiocytosis (histiocytes = macrophages).
It is a rare but rapidly fatal condition if not recognized and treated promptly.
Part 1: The Basic Science (Pathophysiology)
Normal Cytotoxic Killing - What Should Happen
When a virus infects a cell, cytotoxic T lymphocytes (CTLs) and NK cells recognize the infected cell and kill it using a two-step process:
- Pack the weapon: They load cytotoxic granules containing perforin (a pore-forming protein) and granzymes (enzymes that trigger apoptosis)
- Deliver it: Granules dock, prime, and fuse with the target cell membrane - perforin punches holes, granzymes enter and kill the cell
Killing the infected cell removes the antigen stimulus → immune response winds down (activation-induced cell death). This is the normal "off switch."
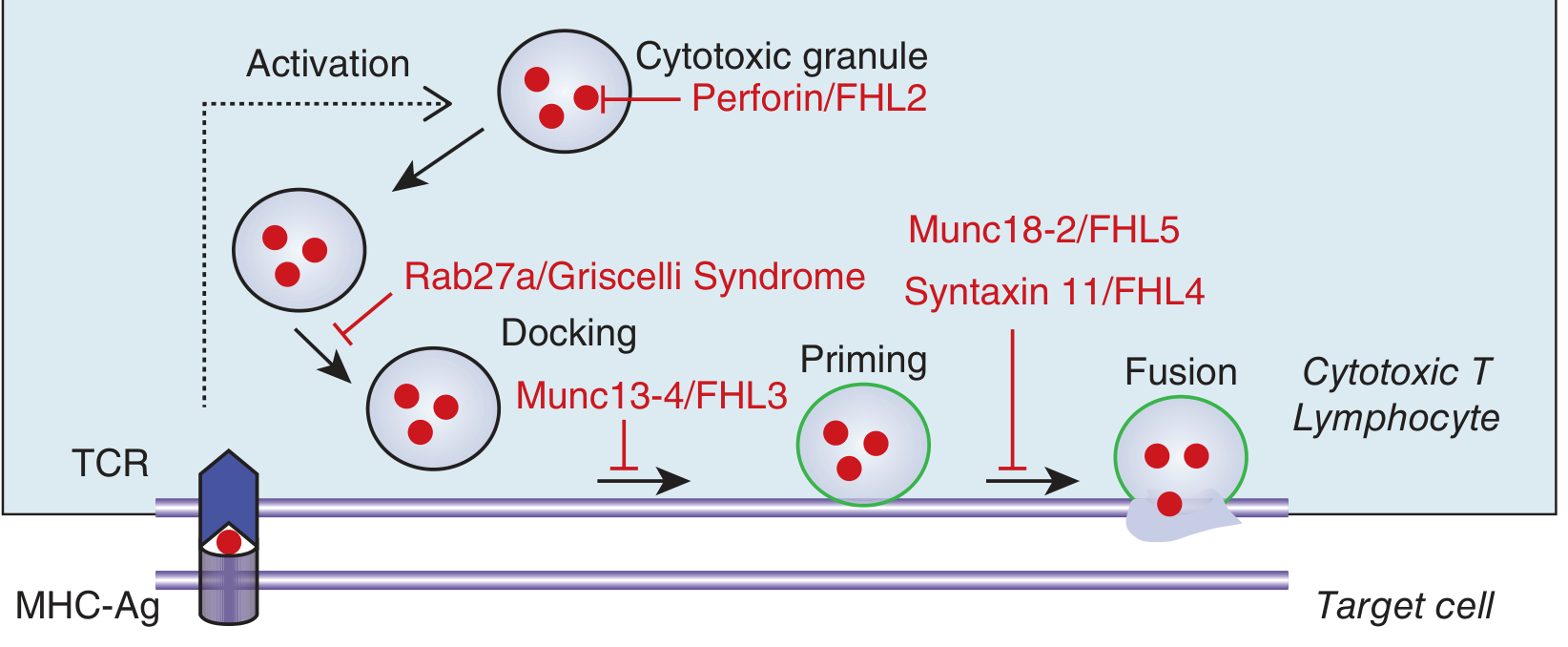
The diagram above shows exactly where each HLH gene mutation blocks the killing machinery.
What Goes Wrong in HLH
When this killing machinery is broken (genetic or acquired), the infected cell cannot be eliminated. The CTLs and NK cells keep trying, but instead of killing the target, they switch to their second effector mode: secreting cytokines, especially IFN-γ (interferon-gamma).
IFN-γ is the master activator of macrophages. Persistently high IFN-γ causes macrophages to become overactivated, leading to:
- Mass production of more cytokines: TNF-α, IL-6, IL-12, IL-10, IL-2 ("cytokine storm")
- Macrophages start phagocytosing healthy blood cells (hemophagocytosis)
- Suppression of normal bone marrow blood cell production
- Systemic inflammation → shock → multiorgan failure
Key cytokines released: IFN-γ, TNF-α, IL-2, IL-4, IL-6, IL-10, IL-12. The result is a systemic inflammatory picture indistinguishable from septic shock. (Robbins Basic Pathology, p. 2806)
Part 2: Classification - Two Broad Types
Primary HLH (Genetic / Familial)
Inherited defects (mostly autosomal recessive) that break the killing machinery at specific steps:
| Gene | Protein | Step Blocked | Name |
|---|---|---|---|
| PRF1 | Perforin | Granule content | FHL2 (~30%) |
| UNC13D | Munc13-4 | Docking | FHL3 (~30%) |
| STX11 | Syntaxin 11 | Priming/Fusion | FHL4 (~10%) |
| STXBP2 | Munc18-2 | Fusion | FHL5 (~20%) |
| RAB27A | Rab27a | Docking | Griscelli Syndrome type 2 |
| LYST | LYST protein | Granule trafficking | Chédiak-Higashi Syndrome |
| SH2D1A | SAP protein | NK/NKT cell signaling | XLP1 (X-linked, EBV-triggered) |
| BIRC4/XIAP | XIAP | Inflammasome regulation | XLP2 (X-linked) |
Associated syndromes with partial albinism: Griscelli syndrome (silvery-gray hair), Chédiak-Higashi (giant lysosomes in leukocytes, progressive neurologic disease), and Hermansky-Pudlak syndrome type II all combine HLH with abnormal pigmentation - hair examination can help in diagnosis.
Epidemiology: Familial HLH affects ~1 in 50,000 live births. Median onset is 3-6 months. It is one of the most common fatal inherited immunodeficiencies alongside SCID. Untreated, median survival is only 1-2 months. (Harrison's 22E, p. 509)
Secondary HLH (Acquired / Non-Genetic)
More common in adults. Caused by external triggers in a genetically predisposed or normal host. Think of it using the "threshold model" - genetic background, underlying inflammation, immunosuppression, and infectious triggers all stack up until a threshold is crossed and HLH ignites.
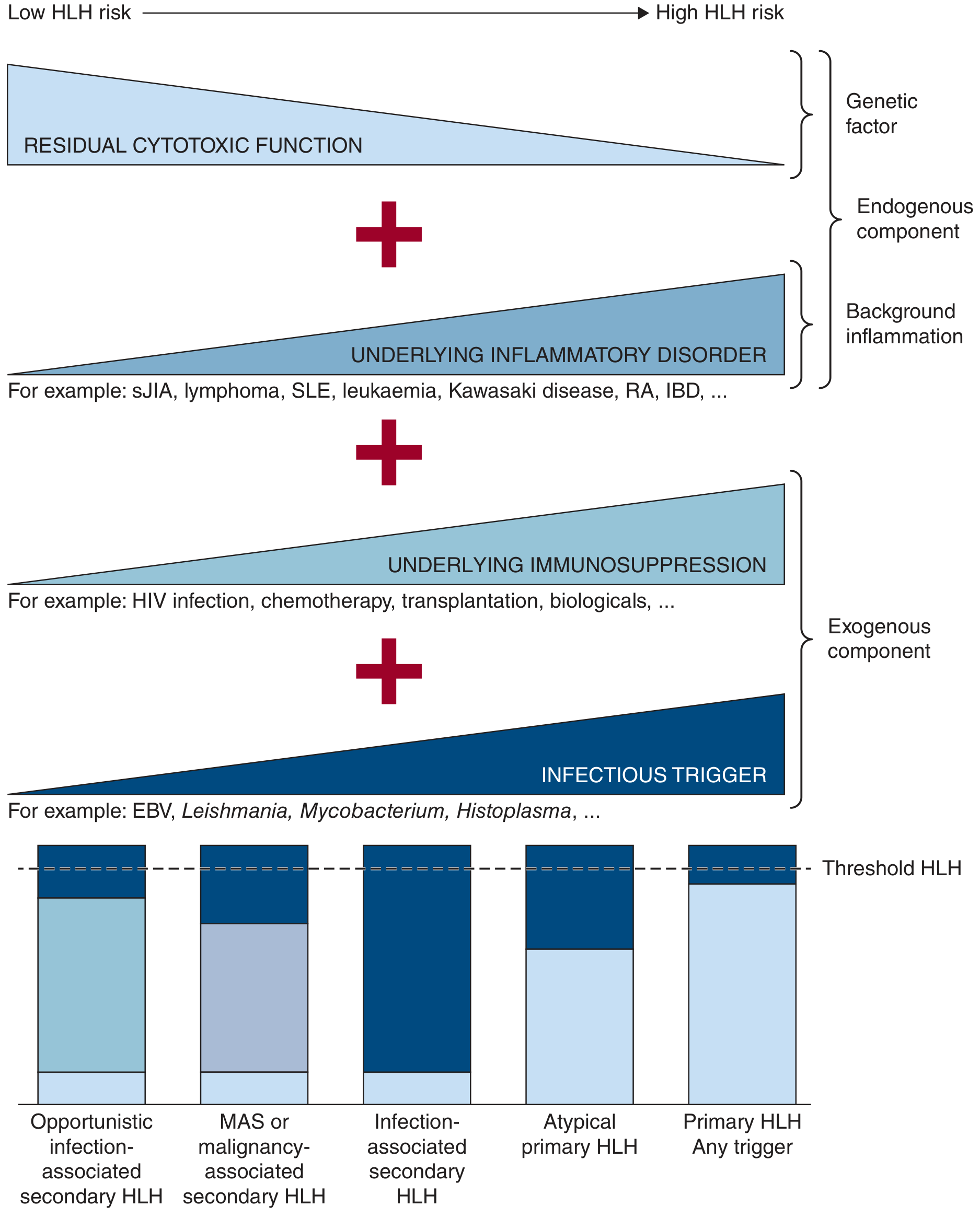
Common triggers in adults:
| Category | Examples |
|---|---|
| Infection (~70% viral) | EBV (most common), CMV, HIV, HHV-8, influenza, parvovirus; also bacteria, fungi, parasites |
| Malignancy | Lymphoma (especially T-cell lymphoma) is the trigger in ~50% of adult cases |
| Autoimmune disease | Systemic JIA, Adult-onset Still's disease, SLE, vasculitis - called Macrophage Activation Syndrome (MAS) in this context |
| Transplant-associated | Post-HSCT or organ transplant |
Note: "MAS" and "secondary HLH" are essentially the same syndrome triggered by autoimmune/rheumatologic diseases. All forms converge on the same clinical picture.
Part 3: Clinical Features - "What Does the Patient Look Like?"
Patients typically present like severe sepsis that doesn't respond to antibiotics. Blood cultures come back negative, but the patient keeps getting worse.
Cardinal Features
| Finding | Mechanism |
|---|---|
| Persistent high fever | IL-1, IL-6, TNF-α |
| Splenomegaly (and hepatomegaly) | Macrophage/lymphocyte infiltration; sequestration |
| Cytopenias (anemia, thrombocytopenia, neutropenia) | Hemophagocytosis + suppressed marrow production |
| Very high ferritin | Macrophage activation; >500 µg/L is criterion, often >>10,000 |
| Elevated triglycerides | TNF-α inhibits lipoprotein lipase |
| Low fibrinogen | Consumed by DIC + suppressed production |
| Elevated liver enzymes | Hepatitis from macrophage infiltration |
| Elevated sIL-2R (sCD25) | Reflects massive T-cell activation |
CNS Involvement (Important!)
About one-third of children with familial HLH have neurologic symptoms at diagnosis: seizures, decreased consciousness, meningeal signs, cranial nerve palsies. CSF is abnormal in ~half. Isolated neurologic presentation (without systemic features yet) can be the first sign in older children/adolescents. (Harrison's 22E)
In Adults
Adults often present with fever + cytopenias + multi-organ failure and are initially thought to have septic shock. Splenomegaly is present in about half. CNS involvement is far less common than in children. Because lymphoma drives ~50% of adult HLH cases, it is easy to miss the underlying malignancy.
Part 4: Diagnosis - The HLH-2004 Criteria
Diagnosis requires either:
A. Molecular confirmation: homozygous/compound heterozygous null mutations in FHL genes
OR
B. At least 5 of 8 clinical/lab criteria:
- Fever
- Splenomegaly
- Cytopenias of ≥2 cell lines (Hgb <9 g/dL, Plt <100×10⁹/L, ANC <1×10⁹/L)
- Hypertriglyceridemia (≥265 mg/dL) and/or hypofibrinogenemia (≤150 mg/dL)
- Ferritin ≥500 µg/L (ferritin >10,000 µg/L has ~90% sensitivity and specificity for HLH)
- Hemophagocytosis on bone marrow biopsy (or in spleen/lymph nodes)
- Low or absent NK cell activity
- Elevated soluble CD25 (sIL-2R) ≥2400 U/mL
Important: Hemophagocytosis on bone marrow biopsy is neither sufficient NOR required to make the diagnosis - it can be absent early in disease and can be seen in other conditions. Do not anchor on this finding alone. (Harrison's 22E)
The HScore (for Secondary HLH in Adults)
A weighted scoring system using 9 variables: fever, organomegaly, immunosuppression history, number of cytopenias, ferritin, triglycerides, fibrinogen, AST, and presence of hemophagocytosis. A score >169 has ~93% sensitivity and ~86% specificity for HLH. Useful especially when some specialized tests are unavailable.
Diagnostic Workup
Full workup should include:
- CBC, CMP, LFTs, ferritin, fibrinogen, triglycerides, sCD25
- Viral studies: EBV, CMV, HIV, HHV-8, hepatitis B/C, HSV, VZV, parvovirus, influenza, adenovirus
- Cultures: blood, urine, CSF
- Bone marrow aspirate + biopsy with flow cytometry and T-cell gene rearrangement
- PET scan and lymph node biopsy to rule out underlying lymphoma before starting therapy - HLH treatment will mask lymphoma and delay its diagnosis
- In children: perforin expression by flow cytometry, NK cell degranulation assay (CD107a), genetic panel
(Goldman-Cecil Medicine, p. 1787)
Part 5: Treatment
The overall approach has two phases: stop the fire then prevent it from restarting.
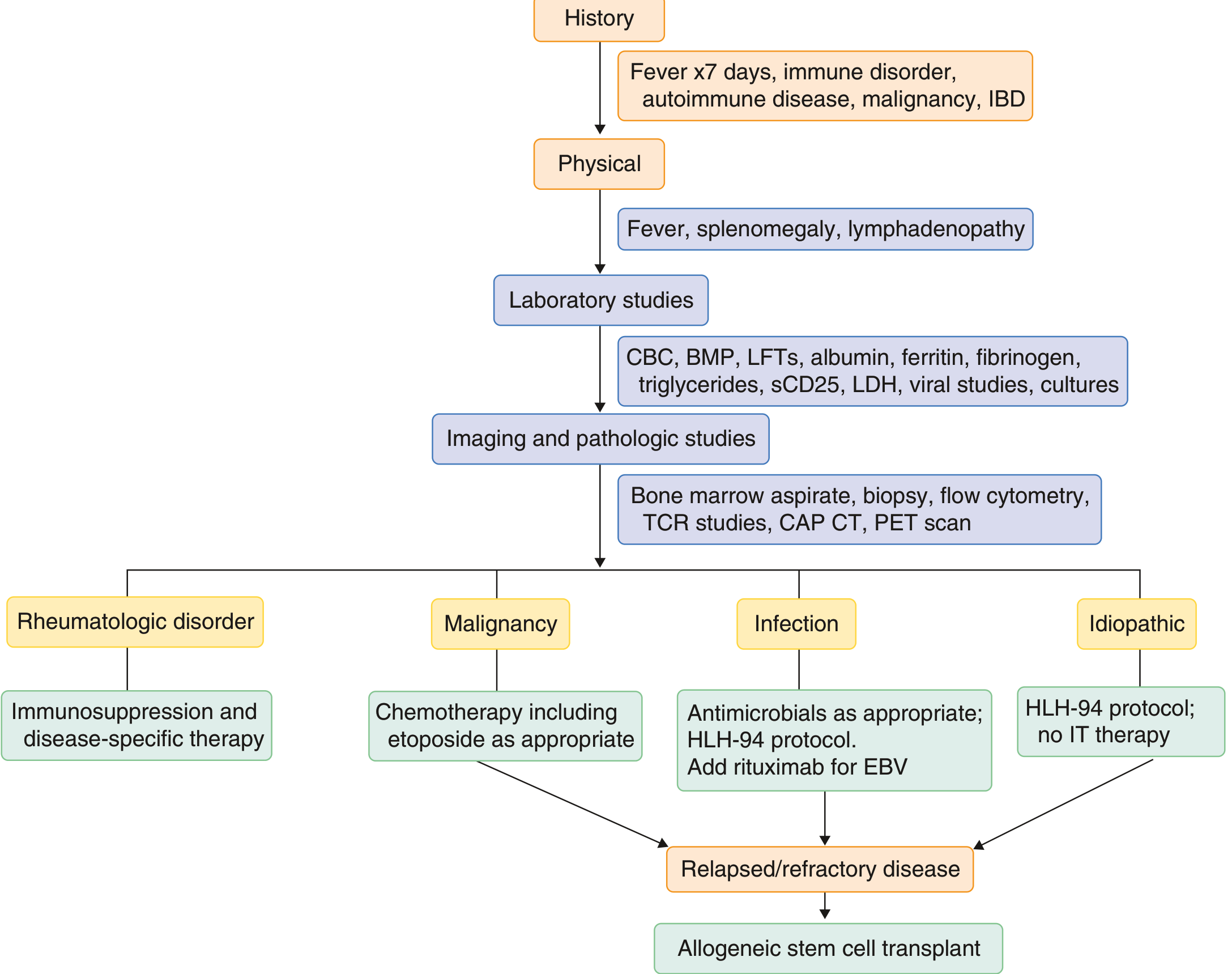
Phase 1 - Control the Hyperinflammation
HLH-94 Protocol (the most established):
- Dexamethasone: 10 mg/m² days 1-14, tapering over 8 weeks (penetrates the BBB well, important for CNS disease)
- Etoposide: 150 mg/m² twice weekly × 2 weeks, then weekly (causes selective depletion of activated T cells and suppresses cytokine production)
- Intrathecal methotrexate: for children with CNS involvement (weeks 3-6)
HLH-2004 modification: adds cyclosporine A from day 1 (blocks T-cell cytokine production)
Newer/salvage agents:
- Emapalumab (anti-IFN-γ antibody): FDA-approved for primary HLH refractory to conventional therapy; blocks the central driver of macrophage activation
- Ruxolitinib (JAK1/2 inhibitor): inhibits multiple inflammatory cytokines; increasingly used for secondary HLH
- Alemtuzumab (anti-CD52): monoclonal antibody that depletes lymphocytes; used in refractory primary HLH
- Rituximab: added for EBV-associated HLH
With modern first-line therapy (etoposide-based), pre-HSCT survival has reached nearly 90% in experienced centers. (Harrison's 22E, p. 509)
Phase 2 - Treat the Underlying Trigger
- Infection: treat the specific infection (antivirals/antibiotics/antifungals); rituximab for EBV-HLH
- Malignancy-HLH: initiate lymphoma chemotherapy promptly - etoposide-containing regimens serve double duty
- MAS/Rheumatologic HLH: disease-specific immunosuppression (e.g., anakinra/IL-1 blockade, tocilizumab/IL-6 blockade for sJIA-MAS, high-dose steroids for SLE)
Phase 3 - Curative Therapy for Primary HLH: HSCT
Allogeneic hematopoietic stem cell transplantation (HSCT) is the only curative treatment for familial HLH. It replaces the defective immune system. Survival after HSCT has reached 85-90% in experienced centers. Overall survival combining first-line therapy + HSCT is now approximately 80%. Without HSCT, familial HLH is uniformly fatal.
For secondary HLH, HSCT is considered in refractory/relapsing disease.
Part 6: Prognosis
| Scenario | Prognosis |
|---|---|
| Familial HLH, untreated | Median survival 1-2 months; uniformly fatal |
| Familial HLH with etoposide + HSCT | ~80% overall survival |
| Asymptomatic siblings (identified by screening) | Close to 100% survival with prophylactic HSCT |
| Adult secondary HLH (no underlying malignancy) | Better prognosis if trigger treated and HLH controlled |
| Malignancy-associated adult HLH | Worst prognosis; depends on treatment of underlying lymphoma |
| Survivors | Risk of renal damage (adults), growth retardation and intellectual disability (children) |
Quick Memory Summary
HLH = immune system can't turn off
→ CTLs/NK cells can't kill infected cells
→ Keep secreting IFN-γ
→ Macrophages overactivated
→ Eat blood cells + release cytokine storm
→ Fever + cytopenias + splenomegaly + organ failure
Diagnose: 5/8 HLH criteria (fever, splenomegaly,
cytopenias, high ferritin, high TG, low
fibrinogen, hemophagocytosis, low NK activity)
Treat: Dex + Etoposide → find trigger → HSCT if primary
Sources:
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 68 - Primary and Secondary HLH
- Robbins & Kumar Basic Pathology, p. 2806-2814
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 2797-2813
- Goldman-Cecil Medicine, Chapter 155, pp. 1787-1792
- Recent review: Recognizing and Managing Secondary HLH in Adults (2025) - PMID 40222878
- 2026 pediatric HLH review: PMID 41481377
This is a shared conversation. Sign in to Orris to start your own chat.