Teach me cystic fibrosis. Give usmle correlations w cystic fibrosis
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
I now have comprehensive, detailed content from multiple authoritative sources. Let me compile the full teaching response.
Cystic Fibrosis - Complete USMLE-Oriented Guide
Sources: Goldman-Cecil Medicine, Robbins Pathology, Murray & Nadel's Respiratory Medicine, Thompson & Thompson Genetics, Murray & Nadel's Respiratory Medicine
1. Overview
Cystic fibrosis (CF) is the most common life-shortening autosomal recessive genetic disease in people of European ancestry. Incidence: ~1:2,500 live births in Northern Europe/Americas (carrier frequency ~1:25). It is caused by absent or reduced function of the CFTR protein (Cystic Fibrosis Transmembrane conductance Regulator).
USMLE Pearl: CF is autosomal recessive. Both parents are carriers (each has a 1:25 chance in European-ancestry populations). Risk of two carriers having an affected child = 1:4.
2. Genetics and Molecular Biology
The CFTR Gene and Protein
- Gene: chromosome 7q31.2, 27 exons, spans ~190 kb
- Protein: ~170 kDa integral membrane protein
- Belongs to the ABC (ATP-binding cassette) transporter superfamily
- The protein has 5 domains:
- 2 membrane-spanning domains (each with 6 transmembrane segments - these form the Cl⁻ channel pore)
- 2 nucleotide (ATP)-binding domains (ATP hydrolysis opens/closes the channel)
- 1 regulatory domain (phosphorylation by PKA activates the channel)
The ΔF508 Mutation
- >70% of all CF patients have at least one copy of F508del (deletion of phenylalanine at position 508)
- This is a Class II mutation - the protein misfolds and is prematurely degraded in the ER (never reaches the cell surface)
- Founder mutation ~52,000 years ago in the Middle East
USMLE Pearl: The most common CF mutation is ΔF508 (delta-F508), a 3-base-pair deletion causing loss of a phenylalanine residue. The protein misfolds and is tagged for destruction before it reaches the apical membrane.
CFTR Mutation Classes (High-Yield Table)
| Class | Defect | Common Mutations | Drug Target |
|---|---|---|---|
| I | No protein synthesis (nonsense/stop codon) | W1282X, G542X | Readthrough agents (ataluren - investigational) |
| II | Misfolding + premature degradation in ER | ΔF508, N1303K | Correctors (lumacaftor, tezacaftor, elexacaftor) |
| III | Protein reaches surface but channel won't open (gating defect) | G551D, G551S | Potentiators (ivacaftor) |
| IV | Protein reaches surface, opens, but conducts Cl⁻ poorly | R117H, R334W | Potentiators |
| V | Reduced amount of normal protein (splicing defect) | 3849+10kbC→T | Potentiators |
| VI | Reduced stability at cell surface | F508del (after correction) | Correctors + potentiators |
USMLE Pearl: Class I-III are severe ("pancreatic insufficient"). Class IV-V are milder ("pancreatic sufficient"). G551D is the classic Class III mutation treated by ivacaftor alone.
3. Pathophysiology
Sweat Glands
Normally, the sweat gland reabsorbs NaCl as sweat travels to the skin surface (CFTR reclaims Cl⁻, Na⁺ follows). In CF, CFTR is absent → sweat chloride ≥ 60 mEq/L (nearly isotonic with plasma). This is the basis of the sweat chloride test.
USMLE Pearl: "Salty baby" - parents notice salty taste when kissing child. This is a classic CF vignette opener.
Airways and Lungs
- Loss of CFTR → reduced Cl⁻ and HCO₃⁻ secretion into the airway lumen
- Simultaneously, Na⁺ absorption increases (via ENaC channels)
- Net result: airway surface liquid dehydration → mucus becomes thick and viscous
- Impaired mucociliary clearance → mucus plugging
- Mucus creates a niche for bacterial biofilms (especially Pseudomonas aeruginosa and Staphylococcus aureus)
- Biofilm organisms activate innate and adaptive immune responses
- Neutrophil elastase causes airway wall destruction → bronchiectasis (predominantly upper lobes)
USMLE Sequence: CFTR mutation → ↓Cl⁻/HCO₃⁻ secretion + ↑Na⁺ absorption → airway dehydration → thick mucus → impaired clearance → chronic Pseudomonas infection → neutrophilic inflammation → bronchiectasis
Pancreas
- CFTR normally secretes bicarbonate- and chloride-rich fluid from pancreatic ductal cells
- Without CFTR → pancreatic duct obstruction → pancreatic autodigestion by retained enzymes
- Exocrine pancreas replaced by fat and fibrous tissue
- 85-90% of CF patients develop pancreatic insufficiency (fat + protein malabsorption)
- Fat malabsorption → deficiency of fat-soluble vitamins: A, D, E, K
- Chronic inflammation can also destroy islet cells → CF-related diabetes mellitus (CFRD) in ~1/3 of patients by age 30
USMLE Pearl: CF causes steatorrhea (bulky, foul-smelling, oily stools) from pancreatic insufficiency. Think fat-soluble vitamin deficiencies: night blindness (A), rickets/osteoporosis (D), hemolytic anemia/neuropathy (E), coagulopathy (K).
Liver
- CFTR absent in bile ducts → thick bile → obstruction of biliary canaliculi → focal biliary cirrhosis
- ~5% progress to hepatic failure requiring transplantation
Small Intestine
- Inadequate hydration of intestinal contents → obstipation, distal intestinal obstruction syndrome (DIOS), intussusception
Reproductive
- Males: >98% are infertile due to congenital bilateral absence of the vas deferens (CBAVD) - the vas deferens fails to develop (obstructive azoospermia, but spermatogenesis is normal)
- Females: Reduced fertility (thick cervical mucus, nutritional/hormonal issues), but some do become pregnant
USMLE Pearl: An otherwise healthy male with infertility and azoospermia on semen analysis but normal testosterone/LH/FSH should prompt CFTR gene testing for CBAVD. This is a forme fruste of CF - can occur with mild CFTR mutations even without pulmonary disease.
4. Clinical Features
Pulmonary
- Chronic productive cough, crackles, wheezing
- Recurrent pulmonary infections
- Bronchiectasis (predominantly upper lobes - differentiates CF from most other bronchiectasis causes which are lower lobe)
- Nasal polyps, chronic sinusitis
- Digital clubbing
- Exacerbations triggered by viral URI
Classic CF pathogens (in order of typical progression):
- Staphylococcus aureus (early childhood)
- Pseudomonas aeruginosa (older children/adults - mucoid form = biofilm)
- Burkholderia cepacia (late disease, very resistant, poor prognosis)
- Stenotrophomonas, Aspergillus, atypical mycobacteria (MALDI)
USMLE Pearl: Mucoid Pseudomonas aeruginosa in the sputum of a young person with recurrent lung infections = think CF until proven otherwise.
Chest X-Ray Findings (Classic on USMLE)
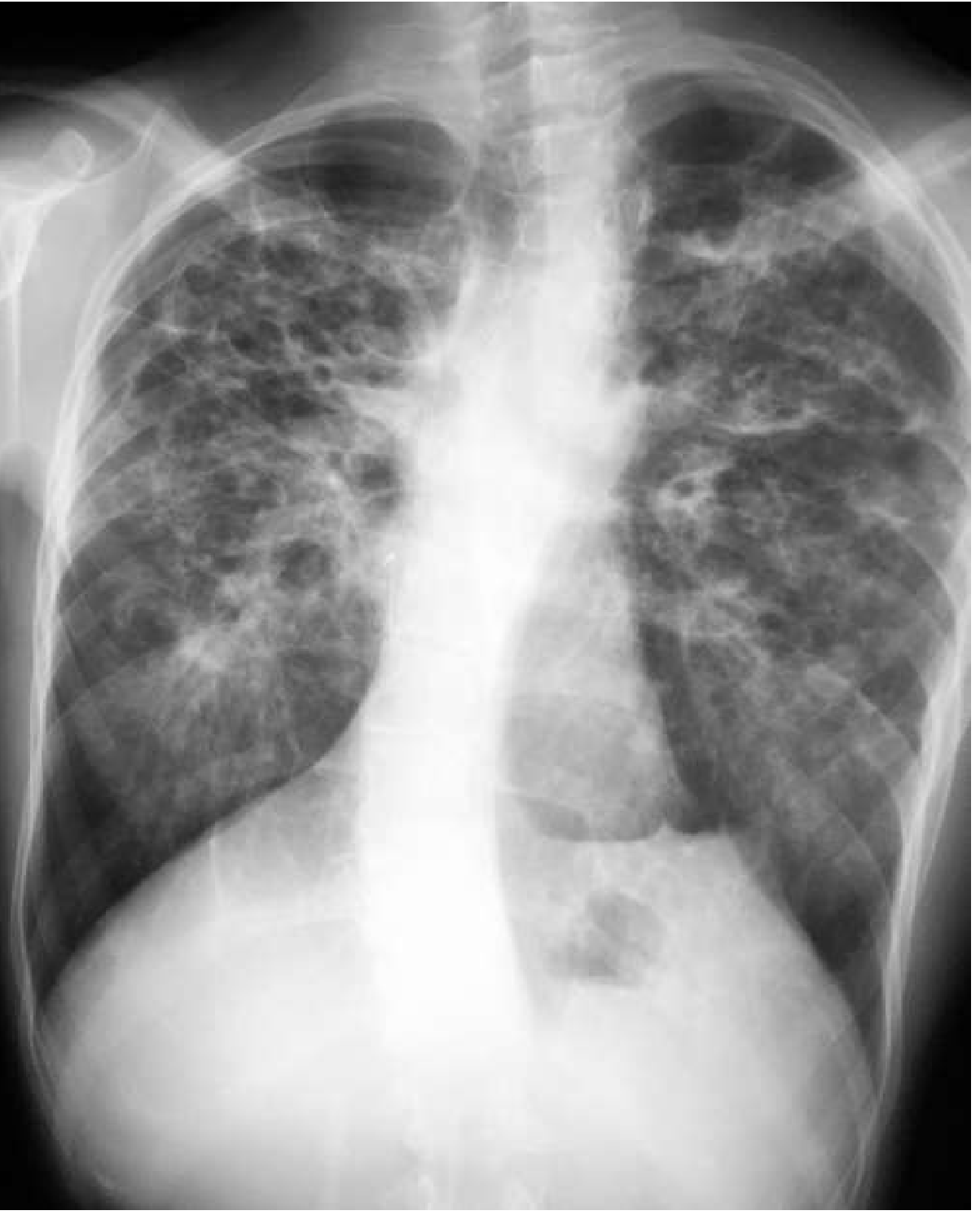
Cystic bronchiectasis preferentially involving both upper lobes with bilateral apical bullae and hilar retraction - Goldman-Cecil Medicine
GI/Nutritional
- Meconium ileus at birth (10-15% of CF newborns) - the classic neonatal presentation; highly specific for CF
- Distal intestinal obstruction syndrome (DIOS) - adult equivalent
- Steatorrhea, failure to thrive, rectal prolapse
- Fat-soluble vitamin deficiencies (A, D, E, K)
- Pancreatic insufficiency → short stature, poor weight gain
Other
- CFRD (CF-related diabetes) - unique insulin-deficient, ketoacidosis-resistant form
- Osteoporosis/osteopenia
- Hypochloremic, hypokalemic metabolic alkalosis (paradoxical: sweat loses NaCl, compensatory renal retention of H⁺ and K⁺ are excreted)
- Erythema nodosum
- Hemoptysis (from bronchiectasis)
USMLE Pearl: A newborn with intestinal obstruction (double bubble or microcolon on X-ray) and no meconium passed within 24-48 hours → meconium ileus → workup for CF.
5. Diagnosis
Newborn Screening
- Immunoreactive trypsinogen (IRT) from heel-stick blood sample - high sensitivity
- Elevated IRT → reflex to CFTR mutation analysis
Sweat Chloride Test (Gold Standard)
- Method: Pilocarpine iontophoresis (pilocarpine stimulates sweat)
- Diagnostic: Cl⁻ ≥ 60 mEq/L
- Normal: Cl⁻ < 30 mEq/L
- Borderline: 30-59 mEq/L (milder variants, "forme fruste")
False-positive sweat tests:
- Nephrogenic diabetes insipidus
- Malnutrition
- Addison disease
- Hypoparathyroidism
- Atopic dermatitis (rarely)
USMLE Pearl: Sweat chloride ≥ 60 mEq/L + compatible clinical features = CF diagnosis confirmed. Two CFTR mutations found on genetic testing also establishes the diagnosis.
Genetic Testing
- Panel testing for ~40 most common CFTR mutations (initial screen)
- Full CFTR sequencing if clinical suspicion high and panel negative
Pulmonary Function Tests
- Obstructive pattern: ↓FEV₁, ↓FEV₁/FVC, ↑RV/TLC (air trapping)
- Progressive FEV₁ decline over time
6. Treatment
Airway Clearance (Cornerstone)
- Chest physiotherapy (postural drainage + percussion)
- Oscillating vest (HFCWO)
- Positive expiratory pressure (PEP) mask
Mucolytic/Secretion-Thinning Agents
- Dornase alfa (rhDNase) - recombinant human DNase breaks down neutrophil-derived DNA in mucus; given by inhalation once daily
- Hypertonic saline (7%) - osmotic hydration of airway surface
- N-acetylcysteine
USMLE Pearl: Dornase alfa (Pulmozyme) improves FEV₁ and reduces exacerbations. The mechanism - neutrophil-derived DNA makes CF mucus viscous; DNase breaks it down.
Infection Management
- Chronic Pseudomonas: Inhaled tobramycin (28 days on/off cycles), inhaled aztreonam, aerosolized colistin
- Exacerbations: IV antibiotics (anti-pseudomonal: piperacillin-tazobactam, ceftazidime, tobramycin, meropenem)
- MRSA: linezolid, vancomycin
- Oral ciprofloxacin for outpatient mild exacerbations
CFTR Modulators (Disease-Modifying - High-Yield)
These drugs fix the underlying CFTR defect rather than just treating symptoms:
| Drug | Type | Mutation Target | How it Works |
|---|---|---|---|
| Ivacaftor (Kalydeco) | Potentiator | Class III (G551D) and others | Keeps CFTR channel open longer |
| Lumacaftor + Ivacaftor (Orkambi) | Corrector + Potentiator | ΔF508 homozygous | Lumacaftor chaperones misfolded protein to surface; ivacaftor then opens the channel |
| Tezacaftor + Ivacaftor (Symdeko) | Corrector + Potentiator | ΔF508 homozygous or compound heterozygous | Similar to lumacaftor/ivacaftor but fewer drug interactions |
| Elexacaftor + Tezacaftor + Ivacaftor (Trikafta) | Triple combo | ΔF508 (1 copy at minimum) | Most efficacious; works for ~90% of CF patients |
USMLE Pearl: Ivacaftor = potentiator (opens the gate). Works for Class III gating mutations (G551D). Lumacaftor/tezacaftor = correctors (help the misfolded protein traffic to the cell surface). Trikafta (elexacaftor/tezacaftor/ivacaftor) = most effective therapy; suitable for patients with at least one ΔF508 allele.
Pancreatic Enzyme Replacement
- Pancrelipase (Creon) - oral lipase/protease/amylase with each meal and snack
- Fat-soluble vitamin supplementation (ADEK)
Other
- Bronchodilators (if airway hyperresponsiveness)
- Anti-inflammatory: ibuprofen (high-dose, slows lung decline in 6-17 year-olds), azithromycin (macrolide - anti-inflammatory + anti-biofilm)
- Management of CFRD: insulin therapy
- Lung transplantation - bilateral sequential transplant for end-stage disease (FEV₁ <30% predicted; hypercapnia; frequent exacerbations)
7. Complications Summary
| System | Complication |
|---|---|
| Pulmonary | Bronchiectasis, pneumothorax, hemoptysis, cor pulmonale, respiratory failure |
| GI | Meconium ileus, DIOS, rectal prolapse, pancreatic insufficiency, distal intestinal obstruction |
| Endocrine | CFRD (by age 30 in ~1/3), osteoporosis |
| Hepatic | Focal biliary cirrhosis, portal hypertension, esophageal varices |
| Reproductive | CBAVD (males, 98% infertile), reduced female fertility |
| Metabolic | Hypochloremic hypokalemic alkalosis, fat-soluble vitamin deficiencies |
8. High-Yield USMLE Correlations
Classic USMLE Scenarios
| Vignette Clue | Think |
|---|---|
| Newborn, no meconium passed in 48h, abdominal distension | Meconium ileus → CF |
| Child with recurrent pulmonary infections + steatorrhea | CF |
| Salty taste when kissing baby | Classic CF clue → sweat chloride test |
| Mucoid Pseudomonas in sputum, young adult, upper lobe bronchiectasis | CF |
| Male infertility, azoospermia, normal testosterone, no sperm in ejaculate | CBAVD → mild CF/CFTR mutation |
| Steatorrhea + failure to thrive + recurrent lung infections in child | CF |
| Sweat Cl⁻ ≥ 60 mEq/L | Diagnostic of CF |
| G551D mutation, on ivacaftor | CFTR potentiator for Class III gating mutation |
| ΔF508 homozygous on Trikafta | Triple CFTR modulator therapy |
| Digital clubbing + chronic cough + upper lobe bronchiectasis | CF |
| Rectal prolapse in child | CF (from chronic straining with fat malabsorption) |
Genetics Pearl for USMLE
- If both parents are carriers (1:25 in whites): 1:4 chance of CF child
- Carrier testing: part of standard prenatal screening
Key Numbers
- Sweat Cl⁻ diagnostic: ≥ 60 mEq/L
- Sweat Cl⁻ normal: < 30 mEq/L
- Most common mutation: ΔF508 (~70% of alleles)
- Pancreatic insufficiency: 85-90% of patients
- Male infertility: >98% (CBAVD)
- Meconium ileus: 10-15% of newborns with CF
- CFRD by age 30: ~33%
9. Prognosis
Median survival has improved dramatically with modern therapy. With CFTR modulators (especially Trikafta), many patients now have near-normal lung function. Historically, death was due to respiratory failure/cor pulmonale. Lung transplant is the definitive option for end-stage disease.
Sources: Goldman-Cecil Medicine 26e (Ch. 77); Robbins & Cotran Pathologic Basis of Disease 10e (Ch. 10); Murray & Nadel's Respiratory Medicine 7e (Ch. 68); Thompson & Thompson Genetics and Genomics in Medicine 9e (Ch. 13)
This is a shared conversation. Sign in to Orris to start your own chat.