Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome" AND pathogenesis
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive, authoritative content from Murray & Nadel, Harrison's, Goldman-Cecil, and Robbins. Here is a thorough synthesis:
Acute Respiratory Distress Syndrome (ARDS): Mechanism
ARDS is a clinical syndrome of acute hypoxemic respiratory failure caused by diffuse alveolar damage (DAD) — a stereotyped lung injury response to a variety of insults. The core mechanism is increased alveolar-capillary permeability leading to non-cardiogenic pulmonary edema, distinct from the hydrostatic edema of heart failure.
Precipitating Insults
ARDS arises from either direct or indirect lung injury:
| Direct Lung Injury | Indirect Lung Injury |
|---|---|
| Pneumonia | Sepsis (most common overall) |
| Aspiration of gastric contents | Severe trauma / multiple fractures |
| Pulmonary contusion | Burns |
| Near-drowning | Pancreatitis |
| Toxic inhalation | Drug overdose, multiple transfusions |
— Harrison's Principles of Internal Medicine, 22nd ed.
Three Sequential Phases

Phase 1 — Exudative (Days 0–7)
This is the core pathophysiological phase and where the primary mechanism plays out.
1. Alveolar-Capillary Barrier Breakdown
The central event is injury to two cell populations:
- Pulmonary microvascular endothelial cells — loss of endothelial barrier integrity is both necessary and sufficient for ARDS development
- Type I alveolar epithelial cells (pneumocytes) — damage disrupts the tight barrier that normally prevents fluid entry into alveoli and also impairs alveolar fluid clearance
Multiple mechanisms drive cell death: necrosis, apoptosis, coagulation activation, and mechanical stretch. The result is a leaky alveolar-capillary membrane through which protein-rich (exudative) fluid floods the interstitium and alveolar spaces.
— Murray & Nadel's Textbook of Respiratory Medicine
2. Neutrophil-Mediated Injury (Central Role)
Proinflammatory cytokines — IL-1, IL-6, IL-8, TNF-α, and lipid mediators (leukotriene B₂) — are released early and drive massive neutrophil recruitment into the pulmonary interstitium and alveolar spaces.
Activated neutrophils cause injury by releasing:
- Neutrophil elastase — degrades extracellular matrix, cleaves surfactant protein A, and injures both endothelium and epithelium
- Reactive oxygen species (ROS) — oxidative damage to cell membranes and proteins
- Matrix metalloproteinases (MMPs) — further matrix degradation
- Myeloperoxidase — generates hypochlorous acid, amplifying oxidative injury
Notably, ARDS has been described even in profoundly neutropenic patients, so neutrophils are major contributors but not the sole mechanism — macrophages and other pathways can sustain injury independently.
— Murray & Nadel's Textbook of Respiratory Medicine
3. Hyaline Membrane Formation
Plasma proteins (fibrin, albumin) that leak into the air spaces aggregate with cellular debris and dysfunctional surfactant to form the characteristic hyaline membranes — the histologic "footprint" of DAD. These whorls line the denuded alveolar walls and impair gas exchange.
4. Surfactant Dysfunction
- Type II pneumocytes are damaged, reducing surfactant synthesis
- Neutrophil elastase degrades surfactant protein A
- The ratio of large (active) to small (inactive) surfactant aggregates falls
- Leaked plasma proteins inhibit surfactant function
The net effect is alveolar instability and collapse, raising surface tension and worsening lung compliance.
5. Microvascular Injury and Pulmonary Hypertension
Concurrent vascular injury causes:
- Microthrombi in pulmonary capillaries (fibrocellular proliferation)
- Hypoxic vasoconstriction
- Compression by positive-pressure ventilation
These reduce blood flow to ventilated regions → increased dead space and pulmonary hypertension.
Physiological Consequences of Phase 1
| Mechanism | Consequence |
|---|---|
| Alveolar flooding + collapse | ↓ FRC, ↓ compliance, intrapulmonary shunt → hypoxemia |
| Microvascular occlusion | ↑ dead space → hypercapnia (late) |
| Pulmonary vascular obliteration | Pulmonary hypertension |
| ↑ work of breathing | Dyspnea → respiratory failure |
Edema predominantly fills dependent lung zones (gravity-dependent), creating a heterogeneous pattern where some alveoli are fluid-filled, others atelectatic, and a few remain aerated (the "baby lung" concept).
Phase 2 — Proliferative (Days 7–14)
In survivors, hyaline membranes are reorganized and type II pneumocytes proliferate to re-line denuded alveolar walls. There is prominent interstitial inflammation and early fibrotic changes. Neutrophil numbers decline. Elevated N-terminal procollagen peptide III in bronchoalveolar lavage (BAL) fluid can be detected as early as 24 hours after onset, suggesting fibroproliferation may begin in parallel with — not after — the inflammatory injury.
Phase 3 — Fibrotic (>21 days)
A subset of patients with persistent ARDS (>2 weeks) develop pulmonary fibrosis with:
- Obliteration of pulmonary capillaries
- Deposition of interstitial and alveolar collagen
- Bullae formation
This phase carries high mortality and impairs long-term quality of life in survivors.
CT Correlation
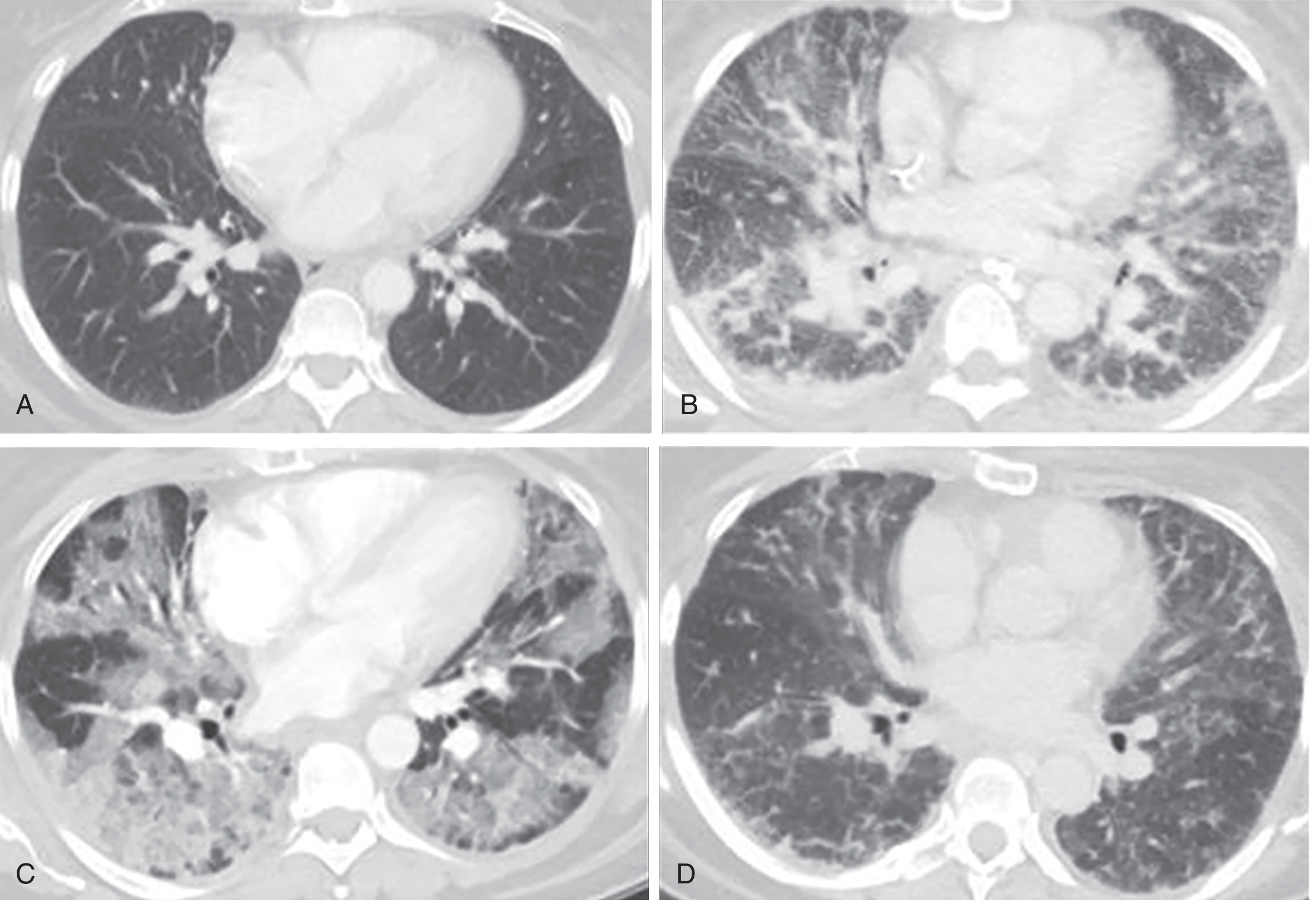
Diagnostic Criteria (Berlin Definition)
| Criterion | Detail |
|---|---|
| Onset | Within 1 week of clinical insult |
| Imaging | Bilateral opacities not explained by effusions, collapse, or nodules |
| Origin | Not fully explained by cardiac failure / fluid overload |
| Oxygenation | Mild: PaO₂/FiO₂ 201–300; Moderate: 101–200; Severe: ≤100 (on ≥5 cmH₂O PEEP) |
2023 proposed updates permit non-intubated patients (high-flow nasal O₂ ≥30 L/min) and allow SpO₂/FiO₂ ratios as an alternative, expanding global applicability.
— Harrison's Principles of Internal Medicine, 22nd ed.
Key Mechanistic Summary
Precipitating insult (sepsis, pneumonia, trauma...)
↓
Systemic/local release of cytokines (TNF-α, IL-1, IL-6, IL-8)
↓
Neutrophil sequestration in pulmonary capillaries
↓
Neutrophil transmigration → alveolar space
↓
Release of elastase, ROS, MMPs
↓
Endothelial + Type I pneumocyte injury/death
↓
↑ Alveolar-capillary permeability
↓
Protein-rich exudate floods alveoli
+ Surfactant dysfunction
+ Hyaline membrane formation
+ Microthrombi
↓
V/Q mismatch, shunt, ↓ compliance → Hypoxemia + Pulmonary HTN
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine, 2-Volume Set
- Harrison's Principles of Internal Medicine, 22nd Edition
- Goldman-Cecil Medicine, International Edition
- Robbins & Kumar Basic Pathology
This is a shared conversation. Sign in to Orris to start your own chat.