Parkisons disease definition etiology epidemology clinical features short pathology with reference
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Parkinson disease"[MeSH Terms] AND epidemiology
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive, authoritative content from multiple textbooks. Let me compile the full answer.
Parkinson Disease
Definition
Parkinson disease (PD) is a chronic, progressive neurodegenerative disorder marked by a hypokinetic movement disorder resulting from selective loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). The resulting dopamine deficiency in the nigrostriatal pathway disrupts motor control in the basal ganglia, producing the clinical tetrad of tremor, bradykinesia, rigidity, and postural instability. PD is classified as the most common primary parkinsonism and the second most common neurodegenerative disease overall (after Alzheimer disease).
The term "parkinsonism" refers to any clinical syndrome sharing these motor features, whether from PD, drugs, toxins, or other neurodegenerative diseases. PD specifically describes the idiopathic, neurodegenerative form with Lewy body pathology.
- Robbins & Kumar Basic Pathology, p. 854
- Goldman-Cecil Medicine, p. [378]
Etiology
PD is believed to result from a combination of genetic susceptibility and environmental factors.
1. Genetic Factors
Most cases (>85%) are sporadic. However, over 20 monogenic forms have been identified and GWAS studies have found >100 risk loci:
| Gene / Locus | Inheritance | Notes |
|---|---|---|
| LRRK2 (PARK8, chr 12q12) | Autosomal dominant | Most common familial cause; G2019S mutation found in up to 5% of Caucasian familial PD; up to 40% of Ashkenazi Jewish and North African Arab cases |
| α-synuclein (SNCA), chr 4q21 | Autosomal dominant | First mutation identified; gene duplications/triplications cause PD; Lewy bodies stain +ve for α-synuclein |
| Parkin (PARK2), chr 6 | Autosomal recessive | Most common inherited defect; causes juvenile-onset PD |
| DJ-1 (PARK7), chr 1 | Autosomal recessive | Early-onset PD |
| PINK1, chr 1p35-36 | Autosomal recessive | Mitochondrial kinase; early-onset |
| GBA (glucocerebrosidase) | Risk factor | Carrier state for Gaucher disease; associated with higher dementia risk and faster progression |
2. Environmental Factors
- MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine): converts to MPP+, which selectively inhibits complex I of the mitochondrial electron transport chain - the strongest evidence for an environmental trigger
- Pesticides and herbicides (rotenone, paraquat): structural homologs of MPTP; higher PD risk in farming, rural living, and well-water consumption
- Heavy metals: manganese, carbon monoxide
- Traumatic brain injury: emergency-department-level TBI associated with increased risk
- Protective factors: caffeine use and cigarette smoking are paradoxically associated with reduced PD risk
3. Pathophysiological Mechanisms
Multiple converging mechanisms are implicated:
-
α-synuclein misfolding and aggregation
-
Mitochondrial dysfunction and oxidative stress
-
Impaired autophagy and lysosomal protein clearance (Parkin, LRRK2, GBA)
-
Excitotoxicity and neuroinflammation
-
Apoptotic cell death
-
Kaplan & Sadock's Comprehensive Textbook of Psychiatry, p. 1718-1719
-
Goldman-Cecil Medicine
-
Robbins & Kumar Basic Pathology, p. 854
Epidemiology
| Parameter | Data |
|---|---|
| Prevalence | ~1 in 1,000 general population; ~1% of those >65 years; 4-5% of those >85 years |
| Incidence | Increases with age in both sexes |
| Sex | Men > Women (ratio ~3:2); incidence consistently higher in males, especially ages 60-79 |
| Age of onset | Typically 55-65 years; uncommon before age 40 |
| Geographic variation | Higher prevalence in European, North American, and Australian populations vs. Asians in the 8th decade; lower reported rates among African Americans (may partly reflect healthcare access) |
| Rank | 2nd most common neurodegenerative disorder after Alzheimer disease |
| Family history | ~15% have a 1st- or 2nd-degree relative with PD |
| Trend | Age-adjusted mortality increased 2.4% per year from 1999 to 2019; prevalence expected to rise sharply with global aging |
A 2024 systematic review and meta-analysis in The Lancet Healthy Longevity confirmed a significant temporal increase in PD prevalence from 1980 to 2023 [PMID: 38945129].
- Goldman-Cecil Medicine
- Textbook of Family Medicine 9e, p. 1249
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry, p. 1717
Clinical Features
Motor Features (Cardinal Tetrad)
1. Tremor
- "Pill-rolling" resting tremor at 4-6 Hz; most prominent at rest, decreases with voluntary movement
- Presenting symptom in ~70% of patients (Hoehn & Yahr data)
- Typically begins asymmetrically - an asymmetric rest tremor is virtually pathognomonic of PD
- Differs from essential tremor (which is bilateral, action-induced, and faster)
2. Bradykinesia
- Slowness of voluntary and spontaneous movement; mandatory for diagnosis
- Manifests as: micrographia (small handwriting), hypophonia (soft voice), reduced arm swing, difficulty with fine motor tasks, slow gait with festination (shuffling short steps)
- Patient often reports "weakness," but strength testing is normal
3. Rigidity
- Increased muscle tone throughout the range of passive motion (unlike spasticity, which is velocity-dependent)
- "Cogwheel" rigidity: ratchety quality felt during passive limb movement (rigidity superimposed on tremor)
- "Lead-pipe" rigidity: smooth, uniform resistance
4. Postural Instability
- Typically a late feature; stooped flexed posture
- Positive pull test (patient falls backwards when pulled)
- Festinant gait with freezing episodes
- Major cause of falls and fractures
Other Motor Features
- Masked facies (hypomimia) - reduced facial expression
- Reduced blink rate (normal: 12-20/min; PD: 5-10/min) with characteristic stare
- Hypophonia and monotonous speech; dysphagia (82% on objective testing)
- Sialorrhea (drooling due to reduced swallowing, not excess saliva production)
Non-Motor Features
Non-motor symptoms often precede motor onset by years (the prodromal phase - up to 20 years; Braak hypothesis):
| System | Symptoms |
|---|---|
| Autonomic | Orthostatic hypotension, constipation, urinary urgency/nocturia, erectile dysfunction, hyperhidrosis |
| Sleep | REM sleep behavior disorder (RBD) - a key prodromal marker; excessive daytime somnolence |
| Sensory | Anosmia/hyposmia (often the earliest symptom), pain, paresthesias |
| Neuropsychiatric | Depression, anxiety, apathy (common early features); hallucinations (later, often drug-related) |
| Cognitive | Mild cognitive impairment in ~30% at diagnosis; up to 80% develop dementia over the course of disease |
Disease Progression
-
Typically progressive over 10-15 years
-
Initially responds to L-DOPA; over time, motor fluctuations ("wearing off," "on-off" fluctuations) and L-DOPA-induced dyskinesias emerge
-
Death commonly from aspiration pneumonia or trauma from falls
-
Adams and Victor's Principles of Neurology, 12th ed., p. 1085
-
Textbook of Family Medicine 9e, p. 1249
-
Robbins & Kumar Basic Pathology, p. 854
-
Goldman-Cecil Medicine
Pathology
Gross Pathology
- Pallor of the substantia nigra (loss of neuromelanin-containing dopaminergic neurons) - the most characteristic macroscopic finding
- Pallor of the locus coeruleus is also seen
Microscopic Pathology
- Neuronal loss of pigmented catecholaminergic neurons in the SNpc with reactive gliosis
- Approximately 60-80% of SNpc neurons must be lost before classic motor symptoms emerge
- Lewy bodies: the pathognomonic inclusion - single or multiple cytoplasmic, round to elongated, eosinophilic inclusions with:
- Dense eosinophilic core surrounded by a pale halo
- Composed of fine filaments of α-synuclein, neurofilaments, and ubiquitin on electron microscopy
- Lewy neurites: dystrophic neurites also containing aggregated α-synuclein
- Pathologic changes extend beyond the SNpc to: brainstem nuclei (locus coeruleus, raphe, dorsal vagal nucleus), olfactory bulb, enteric nervous system, and cortical regions (explaining non-motor symptoms)
Braak Staging (Pathological Progression)
PD neuropathology begins in the lower brainstem and olfactory system (Stage 1-2), progressing rostrally to the SNpc (Stage 3-4), and finally to the neocortex (Stage 5-6). This explains why non-motor symptoms (constipation, anosmia, RBD) precede motor symptoms by years.
Pathogenesis Summary
PD belongs to the family of synucleinopathies (along with DLB and MSA). The key pathogenic cascade involves:
- Abnormal folding of α-synuclein → oligomeric and fibrillar aggregates
- Impaired autophagy and lysosomal clearance (mutations in Parkin, LRRK2, GBA)
- Mitochondrial dysfunction (complex I inhibition)
- Oxidative stress → dopaminergic neuronal death
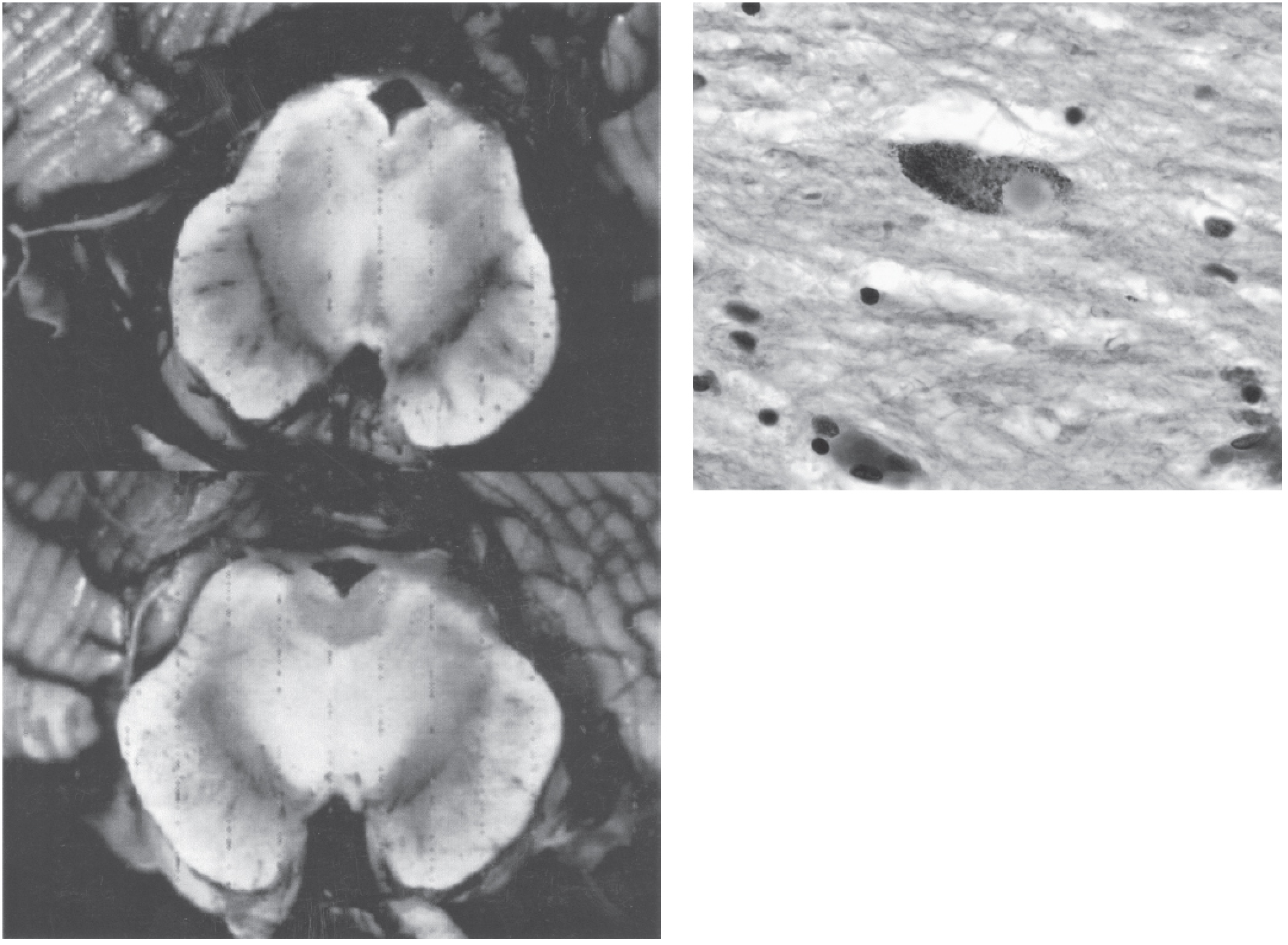
- Robbins & Kumar Basic Pathology, p. 854-855
- Goldman-Cecil Medicine
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry, p. 1717-1718
References
- Robbins & Kumar Basic Pathology (Robbins Pathology), Chapter 21 - Nervous System, "Parkinson Disease," pp. 854-855
- Goldman-Cecil Medicine International Edition, 2 Volume Set - Chapter 378, "Parkinsonism," pp. [3724-3800]
- Bradley and Daroff's Neurology in Clinical Practice - Chapter 38, "Parkinson Disease and Related Disorders"
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry - Section 2.6, "Parkinson Disease / Dementia in Parkinson Disease," pp. 1717-1720
- Adams and Victor's Principles of Neurology, 12th Edition - Chapter 38, "Clinical Features" of Parkinson Disease, p. 1085
- Textbook of Family Medicine, 9th Edition - Chapter: "Parkinson Disease," pp. 1249-1252
- Zhu J et al. "Temporal trends in the prevalence of Parkinson's disease from 1980 to 2023: a systematic review and meta-analysis." Lancet Healthy Longev. 2024 Jul. PMID: 38945129
Give a assessment of physiotherapy
Asking for Preferences
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Parkinson disease"[MeSH Terms] AND "physical therapy"[MeSH Terms]
Finding Sources
Searching PubMed
"Parkinson disease" AND "exercise" AND rehabilitation
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
I now have comprehensive content. Let me compile the full structured answer.
Physiotherapy Assessment in Parkinson Disease
Physiotherapy assessment in PD is a structured, multi-domain evaluation aimed at identifying functional deficits, quantifying disease severity, and guiding individualized rehabilitation. It combines standardized disease-specific scales, functional performance tests, and systems-based physical examination.
1. Purpose of Physiotherapy Assessment in PD
| Goal | Rationale |
|---|---|
| Baseline measurement | Document current motor and functional status |
| Goal setting | Identify impairments most limiting daily life |
| Treatment planning | Target appropriate physiotherapy interventions |
| Monitoring progress | Track response to therapy and disease progression |
| Fall risk stratification | PD is a major fall risk condition |
| Non-motor symptom screening | Many non-motor features interact with rehabilitation outcomes |
2. Subjective Assessment (History)
Chief Complaints
- Tremor (rest vs. action, which limb first, symmetry)
- Stiffness/rigidity (morning stiffness, on/off fluctuations)
- Slowness of movement (bradykinesia)
- Falls and near-falls (frequency, circumstances, injuries)
- Gait disturbances (shuffling, festination, freezing of gait)
- Balance difficulties
Functional History
- Activities of daily living (ADL) status: dressing, hygiene, writing, cooking
- Work and community participation
- Mobility aids used (cane, walker, wheelchair)
- History of falls in the past year
- Impact of medication "on" vs. "off" states on function
Medication History
- Current dopaminergic therapy (levodopa/carbidopa, dopamine agonists)
- Timing of doses relative to assessment - assess ideally during "on" phase
- Motor fluctuations: wearing-off, on-off, dyskinesias
Medical and Social History
- Duration and onset pattern of symptoms
- Cognitive status, anxiety, depression (affect compliance and learning)
- Home environment (stairs, grab rails, floor surfaces)
- Caregiver support
3. Objective Assessment
A. Disease Severity Staging
Hoehn and Yahr Scale - widely used staging system:
| Stage | Description |
|---|---|
| I | Unilateral involvement only; minimal or no functional impairment |
| II | Bilateral or midline involvement; without impairment of balance |
| III | First sign of impaired righting reflex; mild-moderate disability; still independent |
| IV | Fully developed, severely disabling; patient still able to walk/stand unassisted |
| V | Confinement to bed or wheelchair unless aided |
- Bradley and Daroff's Neurology in Clinical Practice
MDS-UPDRS (Movement Disorder Society - Unified Parkinson's Disease Rating Scale)
- Gold standard comprehensive scale for clinical and research use
- 4 parts; total score 0-203 (higher = more severe)
- Part I: Non-motor experiences of daily living
- Part II: Motor experiences of daily living
- Part III: Motor examination (0-132) - most relevant for physiotherapy
- Part IV: Motor complications (dyskinesias, fluctuations)
B. Motor Assessment
Tremor
- Observe at rest (hands in lap), with sustained posture, and during action
- Note: 4-6 Hz pill-rolling rest tremor is characteristic of PD
- Differentiate from essential tremor (action tremor, bilateral, no bradykinesia)
Rigidity
- Passive range of motion testing of wrist, elbow, knee, ankle
- Cogwheel rigidity: ratchety resistance superimposed on tremor
- Lead-pipe rigidity: smooth, uniform resistance throughout range
- Activated rigidity: ask patient to perform contralateral voluntary movement to unmask subtle rigidity
Bradykinesia
- Finger tapping test (rapid alternating movements - note decrement in amplitude/speed)
- Hand opening/closing test
- Foot tapping test
- Assess facial expression (hypomimia), blink rate (reduced to 5-10/min in PD)
- Micrographia assessment (ask patient to write a sentence)
Postural Instability
- Pull Test (Retropulsion Test): Stand behind patient, give a firm pull backward on shoulders - normal = 1-2 steps to recover; abnormal (positive) = multiple steps, no step, or fall
- Positive pull test = Hoehn and Yahr Stage III or above
C. Balance Assessment
Berg Balance Scale (BBS)
- 14 tasks scored 0-4 each; max score 56
- Score <45 predicts fall risk
- Assesses static and dynamic balance
Mini-BESTest (Balance Evaluation Systems Test)
- 14 items; more sensitive to PD-specific balance deficits
- Covers anticipatory postural adjustments, reactive control, sensory orientation, and dynamic gait
- Preferred over BBS for PD
Romberg Test
- Feet together, eyes open then closed - assesses proprioception and vestibular contribution to balance
D. Gait Assessment
Timed Up and Go Test (TUG)
- Rise from a chair, walk 3 meters, turn, return, sit
- Normal <12 seconds; >13.5 seconds = fall risk in PD
- A simple, reliable, widely used measure
10-Metre Walk Test (10MWT)
- Measures comfortable and maximum walking speed (m/s)
- Identifies bradykinetic gait
6-Minute Walk Test (6MWT)
- Assesses functional walking endurance
- Important for cardiovascular exercise capacity assessment
Gait Observation - note:
- Step length (reduced - shuffling)
- Step width (normal early, widened in atypical PD)
- Cadence (rapid, small steps - festination)
- Arm swing (reduced, often asymmetric)
- Trunk posture (stooped/flexed)
- Heel strike (reduced or absent - flat-foot gait)
- Freezing of gait (sudden transient inability to initiate/continue stepping)
- Turns (multiple small steps, en-bloc turning)
E. Posture Assessment
- Observe from anterior, posterior, and lateral views
- Typical PD posture: flexed neck, kyphotic spine, flexed hips/knees, elbow flexion
- Camptocormia (severe anterior trunk flexion) - a specific PD-associated postural deformity
- Pisa syndrome (lateral trunk flexion) - also seen in PD
F. Muscle Strength and Flexibility
- Manual muscle testing (MMT) - note: true weakness is not a primary feature of PD; perceived weakness reflects bradykinesia
- Assess axial and proximal muscle groups (hip extensors, trunk extensors)
- Joint range of motion (ROM) - commonly restricted in shoulders, trunk rotation
- Assess chest expansion (reduced in PD due to rigidity)
G. Respiratory Assessment
- Assess breathing pattern, respiratory rate
- Chest wall rigidity can reduce respiratory function
- Relevant for aspiration risk and cardiovascular exercise capacity
H. Speech and Swallowing (Screen)
- Hypophonia: reduced vocal volume (refer to LSVT® - Lee Silverman Voice Treatment)
- Dysarthria: slurred, monotonous, rapid speech (tachyphemia)
- Dysphagia screening (subjective: eating difficulty, drooling, coughing with meals)
- Refer to Speech-Language Therapist if dysphagia suspected; objective measures: videofluoroscopy, FEES
I. Cognitive and Neuropsychiatric Screening
- Montreal Cognitive Assessment (MoCA): brief, validated, sensitive to PD-associated cognitive impairment (normal ≥26/30)
- Mini-Mental State Examination (MMSE): less sensitive for PD cognitive deficits (misses executive dysfunction)
- Screen for depression (Beck Depression Inventory, GDS)
- Cognitive impairment affects ability to learn new movement strategies and compliance with home programs
J. Functional Independence / ADL
- Parkinson's Disease Questionnaire-39 (PDQ-39): validated PD-specific quality of life measure; 39 items across 8 domains (mobility, ADL, emotional well-being, stigma, social support, cognition, communication, bodily discomfort)
- Barthel Index / FIM for broader ADL function
4. Fall Risk Assessment
Falls are a major cause of morbidity and mortality in PD. Key fall risk factors to assess:
| Factor | Assessment Tool |
|---|---|
| Postural instability | Pull test, Mini-BESTest |
| Gait impairment | TUG, 10MWT |
| Freezing of gait | NFOG-Q (New Freezing of Gait Questionnaire) |
| Cognitive impairment | MoCA |
| Orthostatic hypotension | Lying/standing BP |
| Visual impairment | Acuity, contrast sensitivity |
| Medication effects | Review dopaminergic timing |
| Home environment | Environmental safety checklist |
5. Physiotherapy Interventions (Evidence Summary)
Assessment directly informs the following physiotherapy interventions:
| Impairment | Intervention | Evidence |
|---|---|---|
| Bradykinesia/rigidity | Exercise therapy (aerobic, resistance, combined) | Strong: Cochrane meta-analysis 2024 [PMID: 38588457] |
| Balance deficits | Tai chi, balance training, BIG (LSVT BIG) therapy | RCT evidence; tai chi reduces falls in mild-moderate PD (Goldman-Cecil) |
| Gait (freezing) | Visual/auditory cueing (metronome, laser line), treadmill training | Supported by clinical evidence (Harrison's 22E) |
| Postural deformity | Core strengthening, postural re-education, Alexander Technique | Beneficial |
| Cardiovascular deconditioning | Aerobic exercise (cycling, walking) | Both endurance and resistance very beneficial (Goldman-Cecil) |
| Fall prevention | Multifactorial exercise program | Effective across PD stages |
| Speech/voice | LSVT® LOUD | Validated treatment for hypophonia in PD (Scott-Brown's) |
| ADL/occupational | Occupational therapy, task-specific training | Multidisciplinary approach recommended (Neuroanatomy through Clinical Cases) |
A 2024 Cochrane systematic review and network meta-analysis [PMID: 38588457] confirmed that multiple exercise modalities - including aerobic exercise, resistance training, balance training, and multimodal exercise - produce significant improvements in motor function, balance, and quality of life in PD.
6. Documentation and Re-Assessment
- Reassess at each stage transition (especially Hoehn and Yahr Stage I → III → V)
- Reassess after medication adjustments
- Use standardized scales to allow objective comparison over time
- Coordinate with neurologist, occupational therapist, and speech-language therapist as part of multidisciplinary team (MDT) management
References
- Bradley and Daroff's Neurology in Clinical Practice - Chapters 22, 38, 96 (Hoehn & Yahr Scale, UPDRS, PD Epidemiology and Clinical Features)
- Goldman-Cecil Medicine International Edition - Chapter 378, "Parkinson Disease: Treatment" (exercise, tai chi, rehabilitation)
- Adams and Victor's Principles of Neurology, 12th Edition - "Ancillary treatments" in Parkinson Disease (physiotherapy, yoga, speech exercises)
- Neuroanatomy through Clinical Cases, 3rd Edition - Physical, occupational, and speech therapy in PD
- Scott-Brown's Otorhinolaryngology - Lee Silverman Voice Therapy (LSVT)
- Harrison's Principles of Internal Medicine, 22nd Edition - Cueing strategies for freezing gait
- Ernst M et al. "Physical exercise for people with Parkinson's disease: a systematic review and network meta-analysis." Cochrane Database Syst Rev. 2024 Apr. PMID: 38588457
- Gamborg M et al. "Parkinson's disease and intensive exercise therapy - An updated systematic review and meta-analysis." Acta Neurol Scand. 2022. PMID: 34997759
This is a shared conversation. Sign in to Orris to start your own chat.