Explain the mechanism and pathophysiology of preeclampsia and eclampsia from william obstetrics latest edition
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have everything needed. Note: Williams Obstetrics is not in the library, but Creasy & Resnik's Maternal-Fetal Medicine (a similarly authoritative, comprehensive obstetrics/maternal-fetal medicine reference) is available and contains excellent detail on this topic. I'll compose a thorough, well-organized answer based on this content.
Note on sources: Williams Obstetrics is not in the textbook library, but Creasy & Resnik's Maternal-Fetal Medicine: Principles and Practice (the equivalent authoritative reference in maternal-fetal medicine) is available and covers this topic in equivalent depth. The content below is drawn directly from that text.
Preeclampsia and Eclampsia: Mechanism and Pathophysiology
1. Overview and Definitions
Preeclampsia is a pregnancy-specific disorder typically occurring after 20 weeks' gestation, defined by new-onset hypertension (SBP ≥140 or DBP ≥90 mmHg on two occasions at least 4 hours apart) plus either proteinuria (≥300 mg/24h) or end-organ evidence: thrombocytopenia (<100,000/µL), elevated liver enzymes (>2x ULN), new renal insufficiency (creatinine >1.1 mg/dL), pulmonary edema, or new cerebral/visual disturbances.
Eclampsia is the occurrence of new-onset grand mal seizures in a woman with preeclampsia not attributable to other causes. It can occur antepartum (most common), intrapartum, or postpartum.
2. The Two-Stage Model of Pathogenesis
The currently accepted model involves two sequential stages:
Stage 1 - Abnormal Placentation (Preclinical)
In a normal pregnancy, extravillous trophoblasts invade the decidua and the myometrium, completely remodeling the spiral arteries. This physiologic transformation converts them from narrow, high-resistance muscular vessels into wide, low-resistance conduits - capable of increasing blood flow to the placenta tenfold. The inner elastic lamina and smooth muscle are replaced by fibrinoid material, and the vessels become unresponsive to vasoconstrictors.
In preeclampsia, this trophoblastic invasion is shallow and incomplete. The spiral arteries retain their musculoelastic structure, remaining high-resistance vessels. This creates:
- Reduced uteroplacental perfusion
- Placental ischemia and oxidative stress
- Release of vasoactive and antiangiogenic factors into the maternal circulation
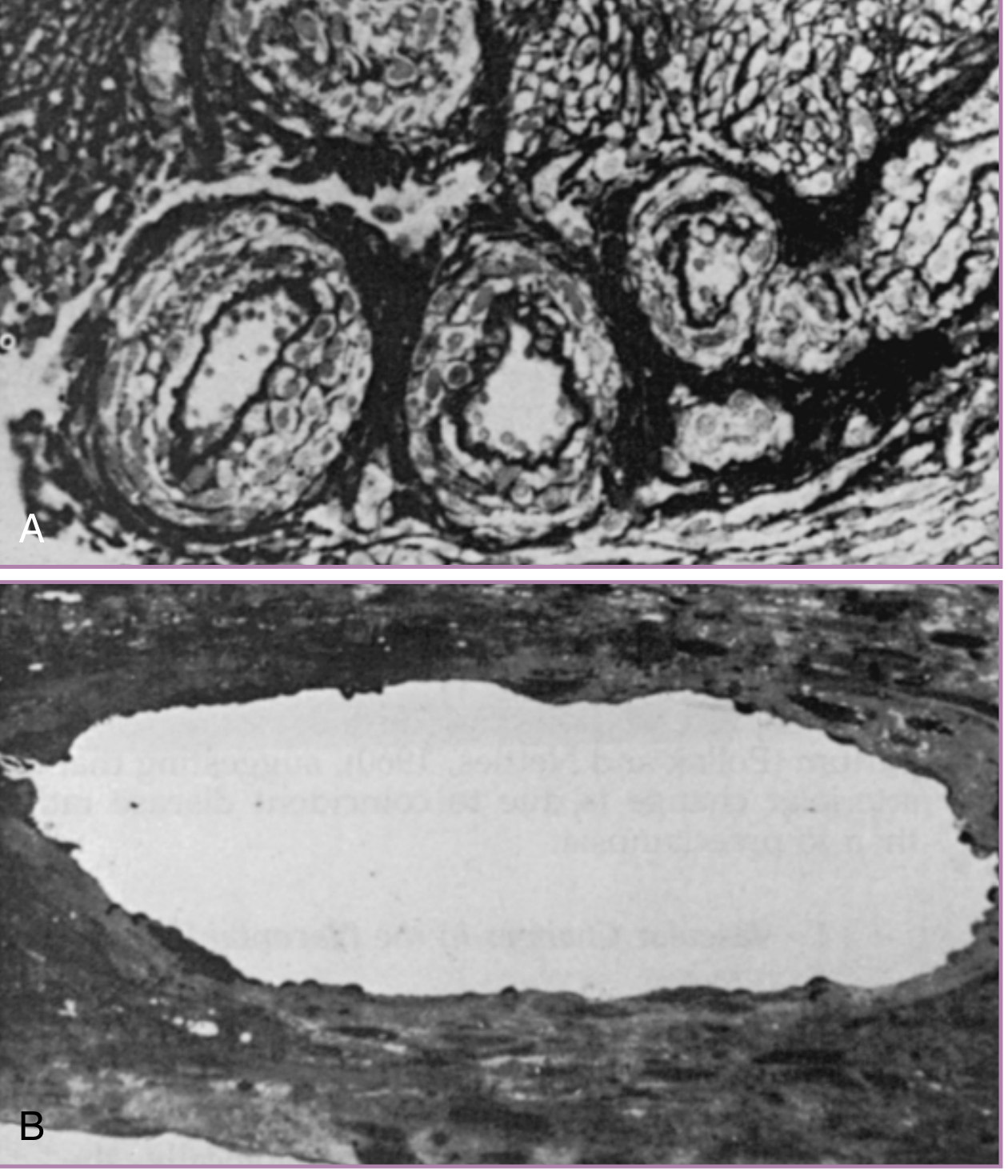
The immunologic basis for failed invasion is important: trophoblasts in normal pregnancy express endothelial-type adhesion molecules (e.g., VE-cadherin, PECAM-1, integrin αVβ3), allowing them to mimic endothelial cells during invasion. This mimicry is absent in preeclampsia. Decidually derived cytokines and local oxygen tension regulate these changes, and complement deposition is found within decidual vessels, suggesting an immunologic component.
Stage 2 - Maternal Syndrome (Clinical Disease)
The ischemic, stressed placenta releases factors into the maternal circulation that cause widespread endothelial dysfunction, triggering the clinical syndrome of hypertension, proteinuria, and multi-organ involvement.
3. The Angiogenic Imbalance - Central Molecular Mechanism
The key discovery linking placental ischemia to maternal endothelial injury is the imbalance between pro-angiogenic and anti-angiogenic factors.
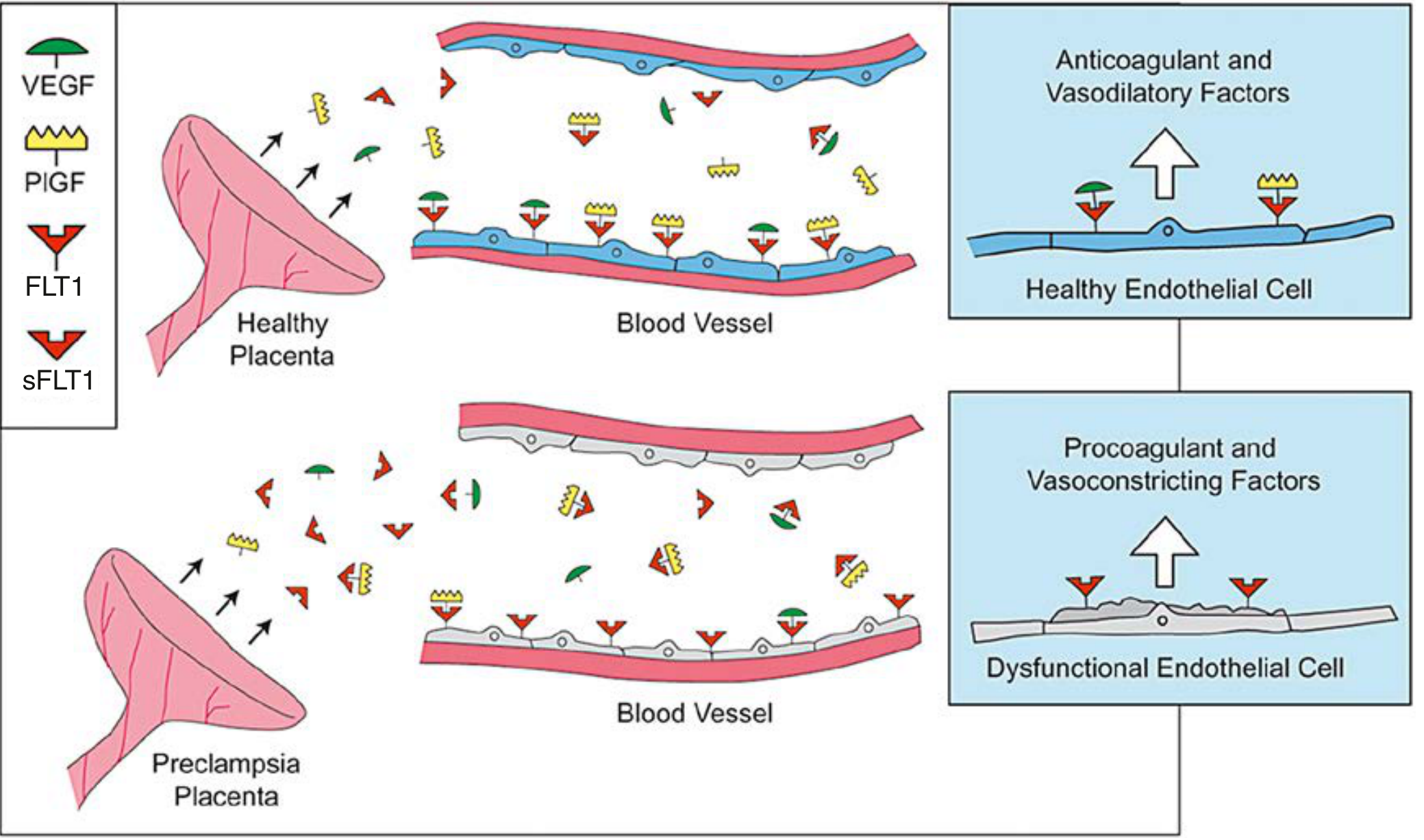
Soluble FMS-like Tyrosine Kinase-1 (sFLT1 / sVEGFR-1)
- sFLT1 is a splice variant of the VEGF receptor FLT1 that lacks the transmembrane and cytoplasmic domains
- The preeclamptic placenta produces greatly excess sFLT1, which circulates freely and acts as a decoy receptor
- It binds and neutralizes both VEGF and placental growth factor (PlGF), preventing them from engaging endothelial surface receptors
- The result: endothelial cells lose trophic VEGF/PlGF support and become dysfunctional
- Circulating sFLT1 rises weeks before clinical symptoms appear; free PlGF falls correspondingly
- When administered experimentally to pregnant rodents, sFLT1 alone induces hypertension, proteinuria, and glomerular endotheliosis - faithfully reproducing the human syndrome
Soluble Endoglin (sEng)
- Endoglin is a co-receptor for TGF-β on endothelial cells
- The preeclamptic placenta releases a soluble form (sEng) that antagonizes TGF-β signaling
- sEng impairs endothelial TGF-β1-mediated vasodilatation and increases vascular permeability
- Combined with sFLT1, sEng worsens the syndrome and may contribute to HELLP
4. Endothelial Cell Dysfunction
This is the central pathologic event linking placental ischemia to all clinical manifestations:
- Prostacyclin (PGI2) production falls - prostacyclin is a potent vasodilator and inhibitor of platelet aggregation produced by healthy endothelium. In preeclampsia, the ratio of thromboxane A2 (TXA2, a vasoconstrictor from platelets) to prostacyclin shifts markedly toward TXA2.
- Nitric oxide (NO) production decreases - NO is a key endogenous vasodilator of pregnancy. Its inhibition dramatically reduces uterine blood flow. In preeclampsia, placental uncoupling of nitric oxide synthase leads to deficient NO production but excess nitrative stress (elevated tissue nitrotyrosine concentrations).
- Procoagulant factors increase - injured endothelium releases von Willebrand factor, fibronectin, and tissue factor
- Vascular permeability increases - fluid extravasation causes edema
- Platelet activation - thromboxane-prostacyclin imbalance drives platelet aggregation and thrombocytopenia
5. Cardiovascular Pathophysiology
Normal pregnancy involves substantial vasodilation and decreased peripheral vascular resistance. In preeclampsia:
- Peripheral vascular resistance increases due to loss of endothelial vasodilators and excess vasoconstrictors (angiotensin II, endothelin, thromboxane)
- Plasma volume contracts - unlike the volume expansion of normal pregnancy, preeclamptic women have reduced circulating volume
- Increased sensitivity to vasopressors - women with preeclampsia show exaggerated blood pressure responses to infused angiotensin II, even before clinical disease (this is used as a prediction test)
- Endothelin-1 levels are markedly elevated and contribute to the hypertension
- The renin-angiotensin-aldosterone system (RAAS) is altered at both systemic and placental levels
6. Renal Pathophysiology
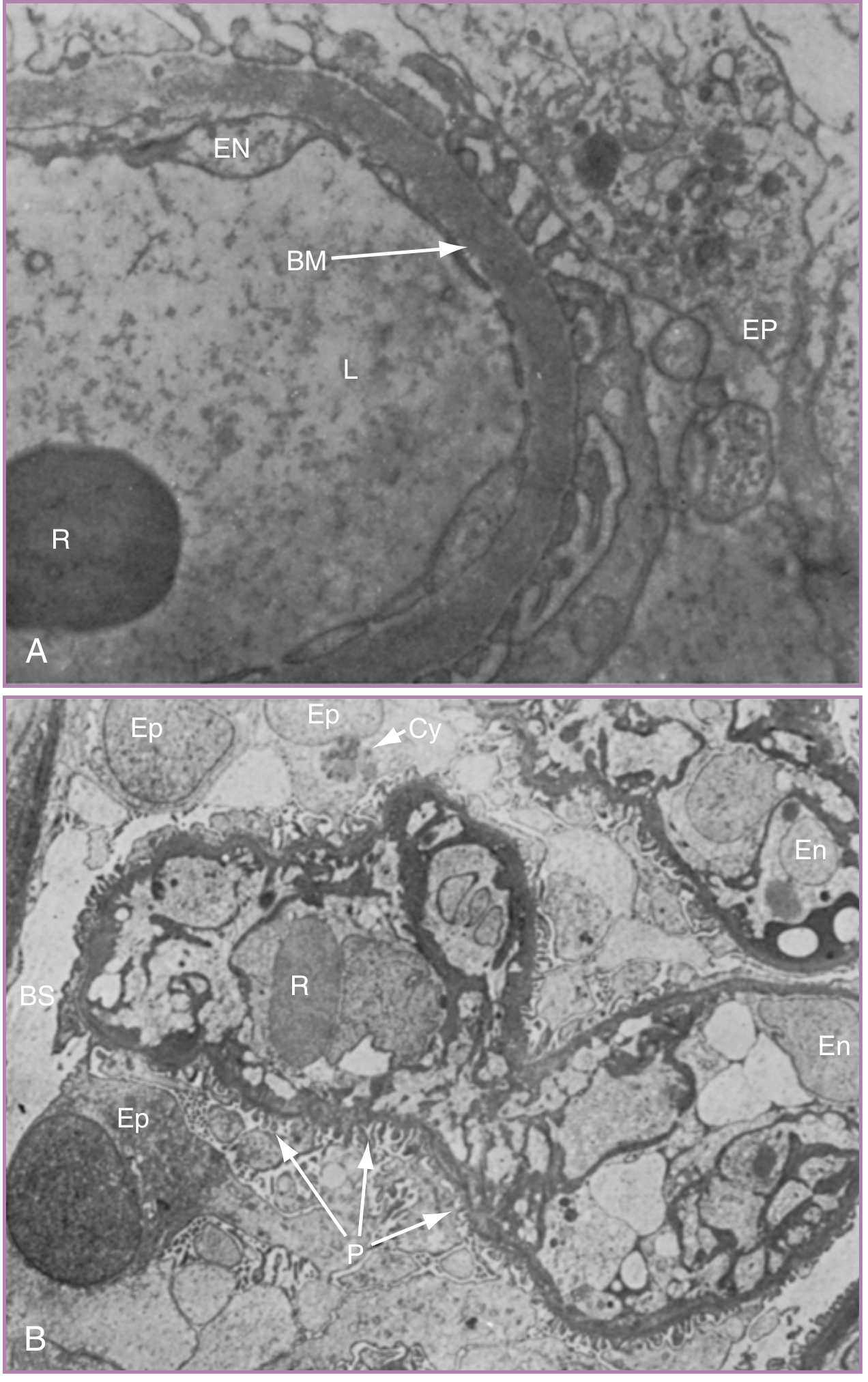
The hallmark renal lesion of preeclampsia is glomerular endotheliosis:
- On electron microscopy, glomerular capillary endothelial cells are markedly swollen (enlarged), with electron-dense inclusions, nearly obliterating the capillary lumen
- The basement membrane is slightly thickened; podocyte foot processes are largely preserved (distinguishing it from primary glomerulonephritis)
- VEGF is required for glomerular endothelial health; sFLT1-mediated VEGF blockade directly causes this lesion
- Consequences: decreased GFR, reduced uric acid and creatinine clearance, proteinuria
The proteinuria in preeclampsia is predominantly due to increased glomerular permeability from endothelial and podocyte injury, not tubular dysfunction.
7. Hepatic Pathophysiology
- Hepatic involvement reflects vasospasm, periportal fibrin deposition, and endothelial damage in hepatic sinusoids
- Periportal necrosis and hemorrhage can occur
- Hepatic capsular distension causes epigastric and right upper quadrant pain (the liver is stretched by subcapsular hematoma or edema)
- Hepatic rupture, though rare, is a catastrophic complication and is an indication for cesarean delivery if delivery is not imminent
- Elevated transaminases (>2x ULN) are a diagnostic criterion for preeclampsia with severe features
- HELLP syndrome (Hemolysis, Elevated Liver enzymes, Low Platelets) represents the severe end of hepatic and hematologic involvement in preeclampsia
8. Hematologic Pathophysiology
- Thrombocytopenia: Platelet activation and consumption at sites of endothelial injury; platelet count <100,000/µL qualifies as severe preeclampsia
- Microangiopathic hemolytic anemia (in HELLP): red blood cell fragmentation on damaged endothelium in small vessels
- Coagulopathy: DIC can occur in severe cases, especially with placental abruption or severe HELLP
- Antithrombin levels fall; thrombin-antithrombin complexes rise, indicating low-grade coagulation activation
9. Neurological Pathophysiology - Eclampsia
The mechanism of eclamptic seizures is not fully understood, but several theories exist:
Cerebral Vasospasm Theory
Generalized vasospasm extends to cerebral vessels. Intense arteriolar constriction leads to focal cerebral ischemia, cytotoxic edema, and neuronal injury - triggering seizure activity.
Cerebral Hyperperfusion / Loss of Autoregulation Theory
Normally, cerebral autoregulation maintains constant cerebral blood flow across a wide range of systemic pressures. In severe hypertension, autoregulation is overwhelmed, leading to breakthrough hyperperfusion, forced dilation of cerebral vessels, and vasogenic edema. This is supported by MRI findings of posterior reversible encephalopathy syndrome (PRES) - characterized by T2/FLAIR hyperintensity predominantly in the posterior parietal and occipital lobes, representing vasogenic edema.
Endothelial Dysfunction Theory
Circulating sFLT1 and other factors damage the blood-brain barrier directly, causing cerebral edema and lowering the seizure threshold.
In practice, both vasospasm and hyperperfusion-related vasogenic edema likely contribute. The clinical features:
- Prodrome: headache (frontal), visual disturbances (photopsia, scotomata, cortical blindness), hyperreflexia, and clonus
- Seizure: typically tonic-clonic, lasting 60-90 seconds
- Postictal phase: confusion, combativeness
- Cerebral hemorrhage (particularly in those with severe hypertension) is the most serious complication and the leading cause of death in eclampsia
10. Placental and Fetal Consequences
- Reduced uteroplacental perfusion from failed spiral artery remodeling causes fetal growth restriction (FGR)
- Placental infarction and abruption are more common
- Oligohydramnios may develop from reduced renal perfusion
- Preterm delivery (iatrogenic or spontaneous) is the most common neonatal complication
11. Immunologic and Genetic Contributions
- Preeclampsia is more common in first pregnancies (nulliparity) and with new paternity (suggesting reduced maternal tolerance to paternal antigens)
- Women with prior prolonged exposure to paternal sperm (longer cohabitation) have lower rates
- Donor egg recipients have a higher risk (the placenta carries fully foreign antigens)
- Natural killer (NK) cell interactions with trophoblast HLA-C molecules at the decidua modulate trophoblastic invasion - abnormal NK cell-trophoblast signaling is implicated
- A strong genetic component exists; family history confers substantially elevated risk, and concordance between maternal and fetal genomes influences susceptibility
12. Summary Cascade
Abnormal trophoblast invasion
↓
Failure of spiral artery remodeling
↓
Uteroplacental ischemia → Oxidative stress
↓
Excess sFLT1 and sEng released into maternal circulation
↓
VEGF/PlGF blockade + TGF-β inhibition
↓
Widespread endothelial dysfunction
↓
↓ Prostacyclin / ↓ NO ←→ ↑ TXA2 / ↑ Endothelin / ↑ Angiotensin II sensitivity
↓
Hypertension + Vasospasm + Platelet activation + ↑ Vascular permeability
↓
Proteinuria (glomerular endotheliosis)
Hepatic injury (periportal necrosis, HELLP)
Thrombocytopenia / microangiopathic hemolysis
Cerebral edema / seizures (Eclampsia)
Placental ischemia → FGR / abruption
Source: Creasy & Resnik's Maternal-Fetal Medicine: Principles and Practice (8th edition), Chapter 45 - Preeclampsia, Eclampsia, and HELLP (pp. 1055-1140). Note: Williams Obstetrics was not available in the textbook library; Creasy & Resnik is the equivalent gold-standard maternal-fetal medicine reference and covers the same pathophysiologic content in comparable depth.
This is a shared conversation. Sign in to Orris to start your own chat.