Hereditary haemorrhagic telengiectasia
Reading File
Finding Sources
Finding Sources
Searching PubMed
"hereditary hemorrhagic telangiectasia"[MeSH Terms]
Reading File
Reading File
Searching PubMed
"hereditary hemorrhagic telangiectasia"[MeSH Terms]
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
I now have comprehensive information to write a thorough answer.
Hereditary Haemorrhagic Telangiectasia (HHT)
Also known as Osler-Weber-Rendu disease, HHT is an autosomal dominant vascular dysplasia characterized by direct arteriovenous connections lacking an intervening capillary bed, affecting skin, mucous membranes, and viscera.
Genetics
HHT is caused by mutations that disrupt TGF-β signalling. The major genetic subtypes are:
| Subtype | Gene | Protein | Locus |
|---|---|---|---|
| HHT1 | ENG | Endoglin | 9q |
| HHT2 | ACVRL1 (ALK-1) | Activin receptor-like kinase 1 | 12q |
| HHT3/4 | GDF2, SMAD4 | BMP9, SMAD4 | Variable |
ENG and ACVRL1 mutations together account for ~85% of cases. SMAD4 mutations cause a combined syndrome of HHT + juvenile polyposis. There are at least five genetic types with hundreds of known mutations. Penetrance is variable but reaches 97% by age 50. - Andrews' Diseases of the Skin, p. 979
Pathophysiology
The basic lesion is a defect in the vessel wall leading to direct arteriovenous connections without a capillary bed. Bleeding results from the mechanical fragility of these abnormal vessels. Both endoglin and ALK-1 are membrane glycoproteins involved in angiogenesis and TGF-β receptor signalling; their loss leads to disordered vessel formation. - Adams and Victor's Principles of Neurology, 12th Ed.
Clinical Features
Diagnosis - Curaçao Criteria (3 of 4 = definite; 2 of 4 = possible)
- Epistaxis - spontaneous, recurrent nosebleeds (most common sign; occurs in 93% of cases)
- Telangiectases - multiple, at characteristic sites: lips, oral cavity, tongue, fingers, nose, nailbeds, palate, ears, soles, palms
- Visceral lesions - GI bleeding; pulmonary, hepatic, cerebral, or spinal AVM
- Family history - one affected first-degree relative
Andrews' Diseases of the Skin, p. 979
Clinical photo - Telangiectasias on the tongue:
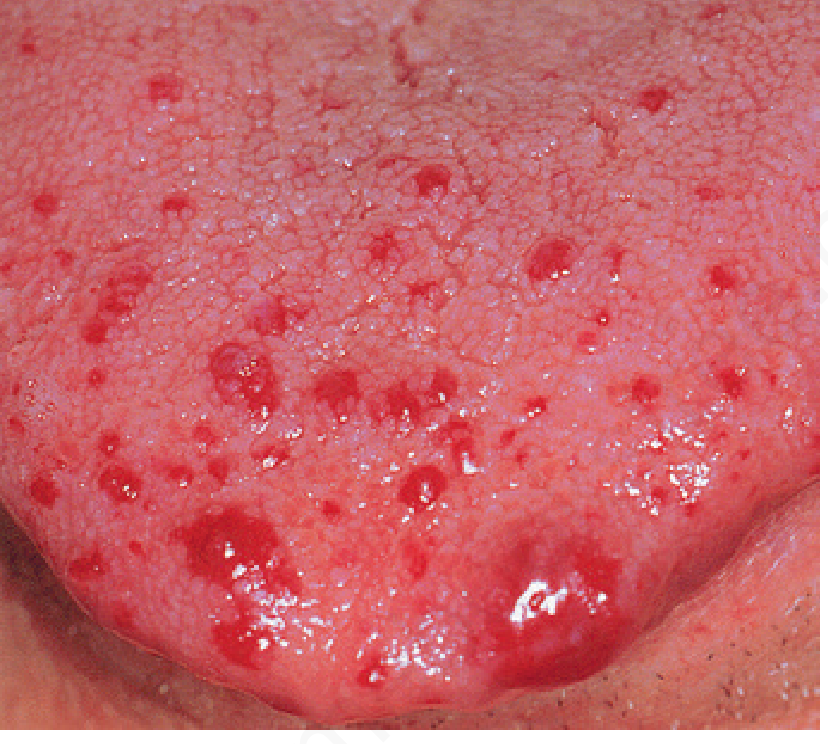
Fig. 35.31 - Hereditary hemorrhagic telangiectasia (Andrews' Diseases of the Skin)
Organ-by-Organ Involvement
Skin and Mucous Membranes
- Small, slightly elevated, bright red to violaceous tufts of dilated capillaries
- First appear at puberty on undersurface of tongue and floor of mouth
- Increase in number during middle age
- Blanch under pressure (distinguishing them from petechiae)
Epistaxis
- The most frequent and persistent sign
- Occurs in 93% of patients
- Worsening epistaxis can herald high-output cardiac failure from hepatic AVMs
Gastrointestinal
- GI bleeding is the presenting sign in up to 25% of patients
- 40-50% develop GI bleeding at some point during the disease
- Chronic iron-deficiency anaemia requiring iron and blood transfusions is characteristic
Pulmonary AVMs
- Can cause haemoptysis and hypoxaemia (right-to-left shunting)
- Major source of paradoxical embolism - predisposing to cerebral abscess and embolic stroke
- Screening recommended even in asymptomatic patients
Hepatic AVMs
- Hepatic artery-to-vein shunting can bypass parenchyma, causing high-output cardiac failure and liver failure
- Liver transplantation may be required in end-stage disease
CNS/Spinal
- Cerebral and spinal AVMs can cause haemorrhage or progressive focal deficits
- Cerebral abscess risk is elevated due to pulmonary AVM-mediated right-to-left shunts (bacteria bypass the pulmonary capillary filter)
- Retinal AV aneurysms occur rarely
Pregnancy
- Can exacerbate HHT; careful monitoring required
Screening
Because of the risk of cerebral abscess and haemorrhage, asymptomatic patients should be screened for pulmonary and cerebral AVMs with imaging. Genetic testing can confirm the diagnosis and guide family screening. - Andrews' Diseases of the Skin, p. 979
Management
Treatment is directed at controlling complications and treating AVMs before they become symptomatic.
Epistaxis
| Approach | Options |
|---|---|
| Local/topical | Packing, cautery, antifibrinolytics (tranexamic acid), topical oestrogens, topical bevacizumab |
| Laser | NdYAG, Argon, KTP532 - laser photocoagulation is popular; reduces telangiectasias |
| Systemic | Systemic or topical oestrogens, tamoxifen, thalidomide (small studies), bevacizumab (IV - conflicting results) |
| Surgical | Septal dermoplasty (replacing mucosa with skin graft), ligation, embolization |
| Last resort | Young's operation (permanent surgical closure of nostrils) - associated with disability but improved quality of life due to reduced bleeding |
GI/Systemic Bleeding
- Iron supplementation and RBC transfusions for chronic anaemia
- Thalidomide and lenalidomide (blocks VEGF) - reduce GI bleeding and transfusion dependence
- Repeated Nd:YAG laser treatments to nasal and GI mucosa
AVMs
- Interventional radiology with selective embolization for pulmonary and CNS AVMs
- Liver transplantation for liver failure or high-output heart failure from hepatic AVMs
Anti-VEGF (Bevacizumab)
- IV bevacizumab has "dramatically improved some severely ill HHT patients," reducing hepatic AVM size and flow, reversing heart and liver failure, and reducing transfusion requirements
- Also used as submucosal injection and topical spray for epistaxis
- Efficacy remains debated for nasal disease specifically - Andrews' Diseases of the Skin, p. 980; Scott-Brown's ORL, Vol 1
Prophylaxis
- Antibiotic prophylaxis for dental and contaminated skin procedures is advocated by some, given the risk of cerebral abscess via pulmonary AVM right-to-left shunting
Differential Diagnosis
- CREST syndrome - mat telangiectases can closely simulate HHT but occur with other features of limited scleroderma (calcinosis, Raynaud's, oesophageal dysmotility, sclerodactyly)
- CM-AVM / CM-AVM2 (capillary malformation-AVM syndromes)
- Generalised essential telangiectasia
- Ataxia-telangiectasia
Key Points Summary
- Autosomal dominant; near-complete penetrance by age 50
- Core genes: ENG (HHT1) and ACVRL1 (HHT2) - together 85% of cases
- Diagnosis by Curaçao criteria (3/4 = definite)
- Epistaxis in 93%; GI bleeding in 40-50% over lifetime
- Screen all patients for pulmonary and CNS AVMs
- Cerebral abscess risk via pulmonary AVM right-to-left shunting is a life-threatening complication
- Bevacizumab (anti-VEGF) emerging as treatment for severe disease
- Large RCT evidence is lacking due to rarity of the condition
Sources: Andrews' Diseases of the Skin (p. 979-980); Scott-Brown's Otorhinolaryngology Head & Neck Surgery Vol 1; Adams and Victor's Principles of Neurology, 12th Ed.; Fitzpatrick's Dermatology
Take refrence from pathology standard books
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Now I have all the content needed from both Robbins books. Let me now compile a comprehensive, well-structured pathology-focused answer.
Hereditary Haemorrhagic Telangiectasia (HHT) - Rendu-Osler-Weber Disease
Primary references: Robbins & Kumar Basic Pathology (10th Ed.) and Robbins, Cotran & Kumar Pathologic Basis of Disease
Classification in Pathology
HHT is classified under Vascular Ectasias - a category of non-neoplastic, permanent dilations of preexisting small vessels (capillaries, venules, arterioles). These are not true neoplasms.
"Telangiectasia is used to describe a permanent dilation of preexisting small vessels (capillaries, venules, and arterioles, usually in the skin or mucous membranes) that forms a discrete red lesion. These can be congenital or acquired and are not true neoplasms."
- Robbins, Cotran & Kumar Pathologic Basis of Disease
Definition
Hereditary Haemorrhagic Telangiectasia (Rendu-Osler-Weber disease) is an autosomal dominant disorder caused by loss-of-function mutations in genes encoding components of the TGF-β signaling pathway in endothelial cells (ECs).
Genetics and Molecular Pathology
| Gene | Protein | Subtype | Notes |
|---|---|---|---|
| ENG | Endoglin | HHT1 | Most common |
| ACVRL1 | ALK-1 (activin receptor-like kinase 1) | HHT2 | Second most common |
| SMAD4 | SMAD4 | HHT + Juvenile Polyposis | Combined syndrome; SMAD4 is a signaling intermediate in TGF-β pathway |
| BMPRIA | Bone morphogenetic protein receptor | Juvenile Polyposis only | TGF-β superfamily |
| Others | - | HHT3/4/5 | At least 5 genetic types known; hundreds of mutations |
Key molecular point: Endoglin and ALK-1 are both membrane-associated proteins involved in angiogenesis through TGF-β signalling in endothelial cells. Their loss causes defective vascular remodelling and the formation of fragile AV connections.
"At least five different genes, most of which modulate TGF-β signaling."
- Robbins, Cotran & Kumar, Chapter 14 (Bleeding Disorders)
SMAD4 and Juvenile Polyposis connection:
"Patients with SMAD4 mutations often have both juvenile polyposis and hereditary hemorrhagic telangiectasia."
- Robbins, Cotran & Kumar, Chapter 17 (GI Pathology)
Pathophysiology
The fundamental lesion is a direct arteriovenous connection without an intervening capillary bed - telangiectasias composed of dilated capillaries and veins, present at birth. The vessel walls are abnormally thin and mechanically fragile due to defective TGF-β-mediated signalling in endothelial cells. Bleeding results from spontaneous rupture of these thin-walled vessels.
"The telangiectasias are malformations composed of dilated capillaries and veins that are present at birth. They are widely distributed over the skin and oral mucous membranes, as well as in the respiratory, gastrointestinal, and urinary tracts. The lesions can spontaneously rupture, causing serious epistaxis, gastrointestinal bleeding, or hematuria."
- Robbins & Kumar Basic Pathology, Chapter 8 (Vascular Ectasias)
Distribution of Lesions
Lesions are present from birth and widely distributed:
- Skin: Face, hands, lips, fingertips, nailbeds, ears, palate
- Mucous membranes: Oral cavity (tongue, floor of mouth - first to appear at puberty), nasal mucosa
- Respiratory tract: Pulmonary AVMs
- GI tract: Stomach, small and large intestine
- Urinary tract: Bladder, kidney
- Liver: Hepatic AVMs
- CNS/Spinal cord: Cerebral and spinal AVMs
Clinical Features (Pathological Basis)
1. Epistaxis
- Most common and persistent sign (93% of patients)
- Due to telangiectasias of the nasal mucosa that rupture spontaneously
- Often the first presenting symptom in childhood/adolescence
2. GI Bleeding
- Second most common bleeding site
- Presenting sign in up to 25% of patients; 40-50% affected at some point in life
- Results in chronic iron-deficiency anaemia (microcytic, hypochromic)
- Requires iron supplementation and blood transfusions in severe cases
3. Hematuria
- From telangiectasias in urinary tract (less common)
4. Arteriovenous Malformations (AVMs)
- Pulmonary AVMs: Right-to-left shunting → hypoxaemia, polycythaemia, paradoxical embolism, cerebral abscess
- Hepatic AVMs: Hepatic artery-to-vein shunting → high-output cardiac failure, portal hypertension, liver failure
- Cerebral/Spinal AVMs: Haemorrhagic stroke, progressive focal deficits
Curaçao Diagnostic Criteria
| Criterion | Description |
|---|---|
| 1 | Epistaxis - spontaneous, recurrent |
| 2 | Telangiectases - multiple, at characteristic sites |
| 3 | Visceral lesions - GI, pulmonary, hepatic, CNS/spinal AVM |
| 4 | Family history - first-degree relative affected |
3 of 4 = Definite HHT | 2 of 4 = Possible HHT
Pathology Context: Where HHT Appears in Robbins
HHT is discussed in three separate contexts in Robbins, which illustrates its multisystem significance:
1. Vascular Pathology (Chapter 8/11)
- Listed under Vascular Ectasias, alongside nevus flammeus and spider telangiectasia
- Emphasises the structural basis of the lesion (dilated capillaries and veins, present at birth, TGF-β signalling defect in ECs)
2. Bleeding Disorders - Vessel Wall Abnormalities (Chapter 14)
- HHT is the most clinically significant cause of bleeding due to vessel wall abnormalities
"Among these conditions, serious bleeding is most often associated with hereditary hemorrhagic telangiectasia."
- Robbins, Cotran & Kumar, Chapter 14
- Context: Placed alongside Henoch-Schönlein purpura, scurvy, Ehlers-Danlos syndrome, and amyloidosis as vascular wall causes of bleeding
3. GI Pathology - Juvenile Polyposis (Chapter 17)
- SMAD4 mutations cause a combined phenotype of HHT + juvenile polyposis
- Juvenile polyposis with HHT carries 30-50% risk of colorectal adenocarcinoma by age 45
4. CNS Tumors / Soft Tissue (mentioned)
- AVMs associated with HHT mentioned among germline mutations predisposing to soft tissue tumours
- Cerebral AVMs in HHT can cause haemorrhagic stroke
Summary Box (Robbins-style)
| Feature | Detail |
|---|---|
| Inheritance | Autosomal dominant |
| Genes | ENG, ACVRL1, SMAD4, BMPRIA, others |
| Pathway | TGF-β signaling in endothelial cells (loss-of-function) |
| Lesion | Dilated capillaries and veins; direct AV connections; no intervening capillary bed; present at birth |
| Key symptom | Epistaxis (most frequent and serious) |
| Bleeding sites | Nasal, GI, urinary, pulmonary, CNS |
| AVMs | Pulmonary, hepatic, cerebral, spinal |
| Complication of pulmonary AVM | Cerebral abscess (paradoxical embolism via right-to-left shunt) |
| Combined syndrome | HHT + Juvenile polyposis (SMAD4) |
| Classification | Vascular ectasia (non-neoplastic) |
Sources: Robbins & Kumar Basic Pathology, 10th Ed. (Chapter 8 - Vascular Disease, p. 336); Robbins, Cotran & Kumar Pathologic Basis of Disease (Chapter 11 - Vascular Disease; Chapter 14 - Bleeding Disorders; Chapter 17 - GI Pathology)
This is a shared conversation. Sign in to Orris to start your own chat.