Bechets disease
Reading File
Finding Sources
Searching PubMed
"Behcet disease" management
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
I now have comprehensive information. Let me compile a complete answer.
Behçet Disease (Adamantiades-Behçet Disease)
Overview
Behçet disease is a systemic, chronic, relapsing inflammatory disorder of unknown etiology classified as a vasculitis affecting all vessel types and sizes - veins, arteries, and capillaries. It is named after Greek ophthalmologist Benediktos Adamantiades (1931) and Turkish dermatologist Hulûsi Behçet (1937). Hippocrates may have described it as early as 460 BC. The condition is considered a "MHC-I-opathy" driven by an abnormal neutrophilic vascular reaction.
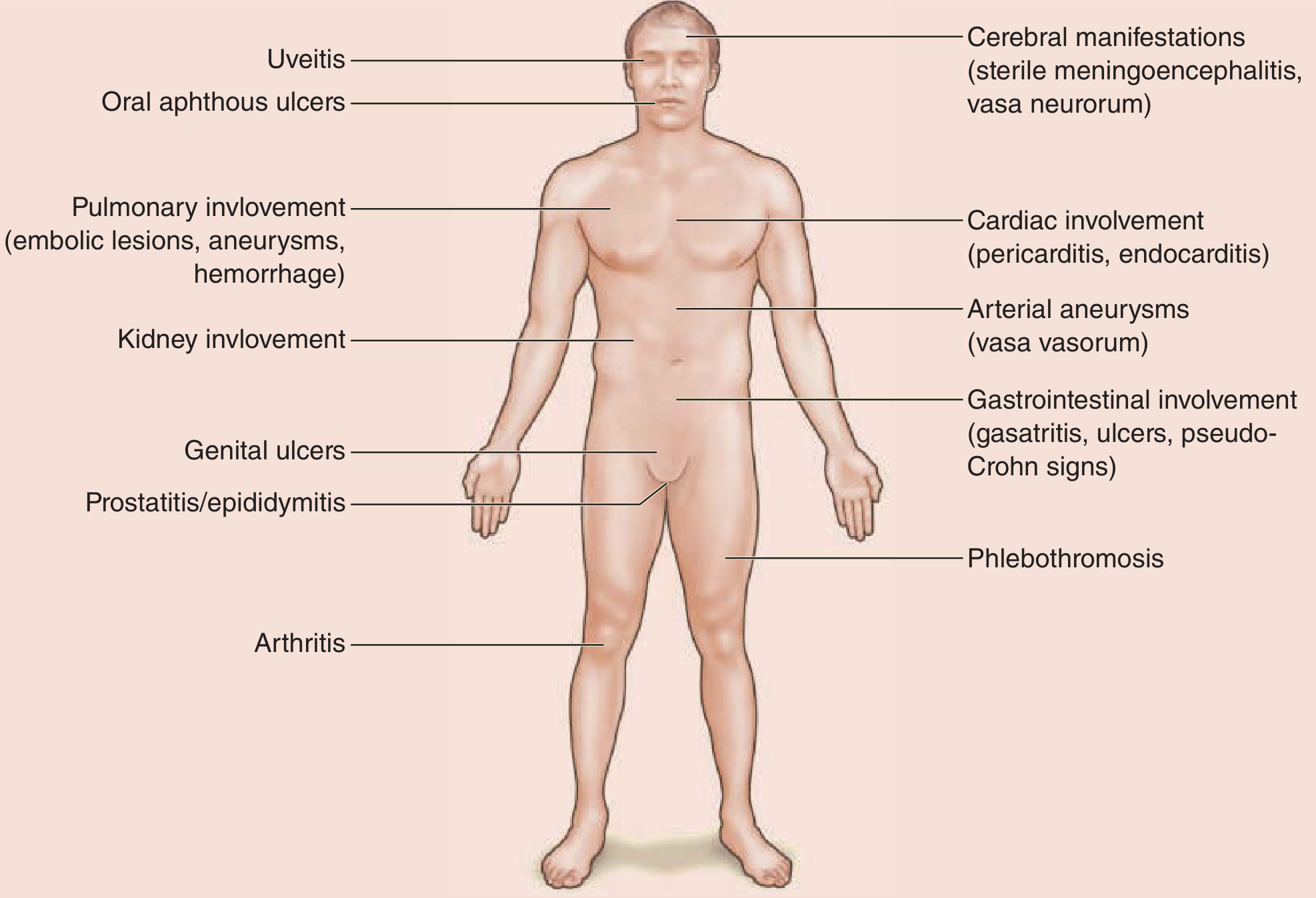
Figure: Behçet disease as a multisystem disorder - Fitzpatrick's Dermatology
Epidemiology
- Endemic along the historical "Silk Road" - highest prevalence in Turkey (80-420 per 100,000), Middle East, Central Asia, and the Mediterranean
- Rare in Northern Europe (0.27-1.18 per 100,000) and the United States (0.75 per 100,000)
- Peak onset in the 20s and 30s; both sexes equally affected overall, though male predominance persists in Arab populations
- HLA-B51 is the strongest known genetic association, linked to more severe disease and ocular involvement; the allele frequency increases toward the east along the Silk Road
Pathogenesis
The etiology is multifactorial:
- Genetic: Strong HLA-B51 association (especially HLA-B5101 and B5108). GWAS studies have identified associations with IL-10, IL-23R-IL12RB2, ERAP-1, CCR1-CCR3, KLRC4, and STAT4 genes. No single Mendelian mode of transmission.
- Infectious triggers: Streptococcal antigens (esp. heat-shock proteins) and herpes simplex virus have been implicated in triggering immune responses via molecular mimicry, though no organism has been proven causative.
- Immune dysregulation: Aberrant neutrophil activation, abnormal T-cell responses (elevated Th1/Th17), and endothelial injury are central mechanisms. Elevated IL-6, IL-8, TNF-α, and IFN-γ are found in active disease.
Clinical Features
Cardinal (Mucocutaneous) Features
1. Oral aphthous ulcers - the most common symptom and usually the first sign
- Recurrent, painful, shallow ulcers on mucous membranes
- Heal without scarring (minor) or with scarring (major/herpetiform)
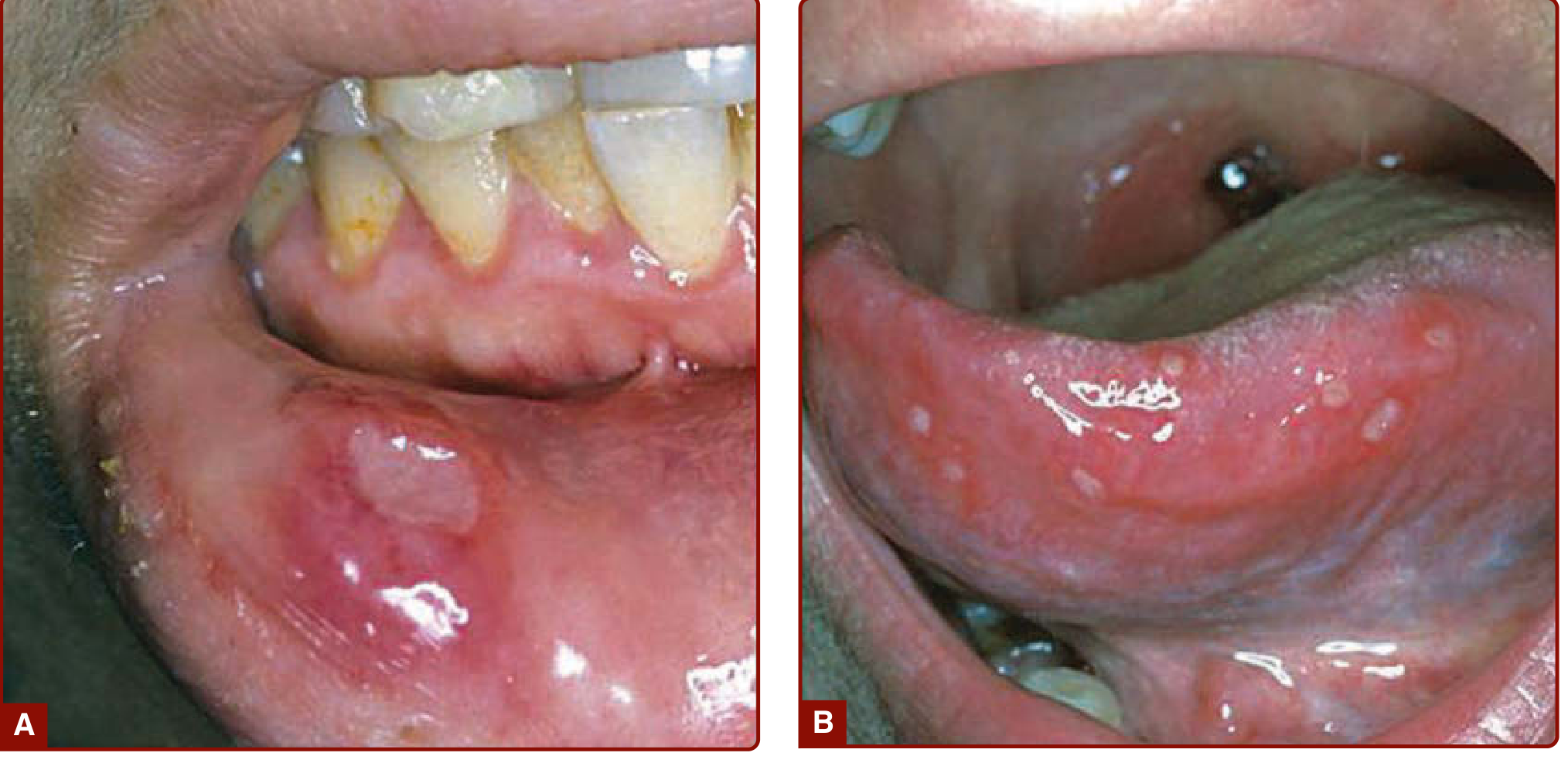
Figure: (A) Single and (B) multiple oral aphthous ulcers - Fitzpatrick's Dermatology
2. Genital ulcers
- Affect the scrotum, penis, vulva, and cervix
- Larger and more painful than oral ulcers; carry a higher risk of scarring (unlike oral ulcers)
- Pathognomonic when combined with oral ulcers
3. Skin lesions
- Pseudofolliculitis, papulopustular lesions
- Erythema nodosum-like lesions (particularly in females)
- Superficial thrombophlebitis
4. Positive pathergy test
- An erythematous papule (>2 mm) or pustule forms within 48 hours at a sterile needle-prick site on the forearm
- Pathergy is more prevalent in patients from the Middle East/Asia than in Western populations
Ocular Involvement
The major cause of morbidity. Posterior uveitis (retinal vasculitis) is the most diagnostically relevant lesion and can lead to blindness if untreated. Other features include anterior uveitis, hypopyon (pus in the anterior chamber), cataract, glaucoma, cystoid macular edema, and ischemic optic nerve atrophy.
Articular Involvement
A nonerosive, asymmetric, seronegative oligoarthritis - typically involves knees and ankles in migratory fashion. HLA-B27-positive erosive sacroiliitis must be excluded.
Vascular Involvement
Both arteries and veins are affected:
- Venous: deep vein thrombosis, cerebral venous sinus thrombosis, Budd-Chiari syndrome
- Arterial: aneurysms (including pulmonary artery aneurysms which can cause life-threatening hemoptysis), occlusions
Neurological Involvement (Neuro-Behçet)
Occurs in ~20% of cases. Manifestations include:
- Aseptic meningitis/meningoencephalitis
- Focal/multifocal neurological deficits from small-vessel ischemic disease (brainstem frequently involved)
- Cerebral venous sinus thrombosis
- CSF: mild pleocytosis, elevated protein
- Psychiatric manifestations: depression, psychosis, dementia
Gastrointestinal Involvement
- Mucosal ulcers, predominantly in the ileocecal region (can mimic Crohn disease)
- GI bleeding, perforation, or obstruction
- Hepatic involvement via Budd-Chiari syndrome
Pulmonary Involvement
- Pulmonary artery aneurysms - a life-threatening complication
- Arterio-bronchial fistulae with massive hemoptysis
Diagnosis
Behçet disease is a clinical diagnosis - no pathognomonic lab test exists.
International Criteria for Behçet Disease (ICBD Scoring System)
| Feature | Points |
|---|---|
| Ocular lesions (recurrent) | 2 |
| Oral aphthosis (recurrent) | 2 |
| Genital aphthosis (recurrent) | 2 |
| Skin lesions | 1 |
| CNS manifestations | 1 |
| Vascular manifestations | 1 |
| Positive pathergy test | 1 (extra) |
Score ≥ 4 = Behçet disease
The older International Study Group criteria (1990) required recurrent oral ulcers PLUS two of: recurrent genital ulcers, ocular lesions, skin lesions, or positive pathergy test.
Investigations
- No specific autoantibodies (ANA, ANCA, RF are negative)
- Elevated ESR, CRP, leukocytosis during flares
- HLA-B51 testing (supportive, not diagnostic)
- MRI brain for neuro-Behçet (hypodense/atrophic changes)
- Angiography for vascular lesions
- CSF analysis if CNS involvement suspected
Differential Diagnosis
| Category | Conditions |
|---|---|
| Oculocutaneous | Erythema multiforme/SJS, Vogt-Koyanagi-Harada, reactive arthritis, pemphigus |
| GI/mucocutaneous | Crohn disease, ulcerative colitis, tuberculosis |
| Articular | SLE, MAGIC syndrome, ankylosing spondylitis |
| Aphthae | Recurrent aphthous stomatitis, herpes simplex, cyclic neutropenia |
| CNS | Multiple sclerosis, Neuro-Sweet disease |
| Genital ulcers | Herpes simplex, Lipschütz ulcer, STIs |
Treatment
Treatment is guided by organ involvement and severity. There is no curative therapy; the goal is remission and prevention of organ damage.
Mucocutaneous Disease
- Topical corticosteroids (triamcinolone mucosal ointment, dexamethasone paste) - first-line for oral/genital ulcers
- Topical anesthetics for pain relief
- Colchicine - effective for oral and genital ulcers, erythema nodosum, arthritis; often the first systemic agent
- Sucralfate suspension (5 mL × 4/day) - reduces oral aphthae frequency and pain
- Thalidomide - useful for isolated refractory mucocutaneous disease
Systemic Disease
| Manifestation | Treatment |
|---|---|
| Uveitis (mild-moderate) | Topical/systemic corticosteroids |
| Uveitis (severe/posterior) | Systemic corticosteroids + azathioprine (first choice); cyclosporine A |
| Neuro-Behçet | High-dose corticosteroids + azathioprine; infliximab for refractory cases |
| Vascular (venous thrombosis) | Corticosteroids; anticoagulation for cerebral venous sinus thrombosis |
| Arterial aneurysms | Immunosuppression (corticosteroids + cyclophosphamide); surgery if needed |
| GI involvement | Prednisone; azathioprine, 6-mercaptopurine |
| Refractory disease | Infliximab (anti-TNF), interferon-α, cyclosporine, methotrexate, chlorambucil |
Key drug notes:
- Azathioprine: One of the first choices for severe uveitis; shown in RCT to prevent new eye disease and protect vision (Yazici et al., NEJM 1990)
- Colchicine: Good evidence for mucocutaneous and joint disease
- Anti-TNF agents (infliximab, adalimumab): Reserved for severe refractory disease, particularly ocular and neurological involvement
- Apremilast: Emerging evidence for oral ulcers
Prognosis
- Clinical course is variable with relapses and remissions
- Severe complications (blindness, meningoencephalitis, hemoptysis, intestinal perforation, severe arthritis) occur in ~10% of patients
- Mortality: 0-6% across ethnic groups
- Poor prognostic markers: HLA-B51 positivity, male sex, early systemic involvement, superficial thrombophlebitis, ocular disease at onset
- Ophthalmic and neurological sequelae are the leading causes of long-term morbidity
- Leading causes of death: CNS involvement, pulmonary/large vessel disease, bowel perforation
Sources: Fitzpatrick's Dermatology (Chapter 141), Bradley & Daroff's Neurology in Clinical Practice, Textbook of Family Medicine 9e, Sleisenger & Fordtran's GI & Liver Disease, Harrison's Principles of Internal Medicine 22e
This is a shared conversation. Sign in to Orris to start your own chat.