What are the errors that can happen in a DNA? b) Which is the most common error in a DNA? Refer lippincot
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I have enough context from the Lippincott Biochemistry textbook and my knowledge of its content to give a thorough, accurate answer. The textbook's DNA Repair chapter (Chapter 30 in 8th ed) covers all these topics in detail. Let me now provide a comprehensive answer based on what Lippincott Illustrated Reviews Biochemistry teaches:
DNA Errors (Mutations) - Lippincott Illustrated Reviews: Biochemistry, 8th ed.
a) Types of Errors (Mutations) in DNA
Lippincott classifies DNA mutations based on the type and scale of the nucleotide change:
1. Point Mutations (Single Base Substitutions)
A single nucleotide is replaced by another. These are the most extensively discussed in Lippincott.
a. Transitions
- A purine is replaced by a purine (A ↔ G), or a pyrimidine by a pyrimidine (C ↔ T).
- Example: In sickle cell disease, GAG → GTG at codon 6 of the β-globin gene (glutamate → valine). This is a transversion (below), but Lippincott uses it as the classic point mutation example.
- Transitions are more common than transversions because the geometry of the base pair is less disrupted.
b. Transversions
- A purine is replaced by a pyrimidine, or vice versa (A/G ↔ C/T).
- Less common; cause a greater structural disturbance.
2. Based on Effect on Protein: Functional Classes of Point Mutations
| Type | Description | Example |
|---|---|---|
| Silent (synonymous) | Codon change but same amino acid (due to degeneracy of the genetic code). No effect on protein. | GCU → GCC (both = Ala) |
| Missense | Codon change results in a different amino acid. Protein may be non-functional or partially functional. | Sickle cell: GAG (Glu) → GTG (Val) |
| Nonsense | Codon change produces a premature STOP codon (UAA, UAG, UGA). Leads to truncated, usually non-functional protein. | CAG (Gln) → UAG (Stop) |
3. Frameshift Mutations (Insertions and Deletions / Indels)
- One or more nucleotides are inserted into or deleted from the DNA sequence.
- This shifts the reading frame of the mRNA downstream of the mutation.
- Nearly all codons after the insertion/deletion are read incorrectly, producing a completely aberrant protein, often with a premature stop codon.
- Deletions of multiples of 3 nucleotides do not cause a frameshift - they just remove one or more amino acids.
- Example: Many cases of Duchenne muscular dystrophy are caused by frameshift deletions in the dystrophin gene.
4. Trinucleotide (Triplet) Repeat Expansions
- A specific 3-base sequence is abnormally repeated many more times than normal.
- Once the number of repeats exceeds a threshold, it causes disease - often with anticipation (worsening in successive generations).
- Examples: Huntington disease (CAG repeats in the huntingtin gene), Fragile X syndrome (CGG repeats), Myotonic dystrophy (CTG repeats).
- Lippincott notes that Southern blotting can detect trinucleotide repeat expansions.
5. Large-Scale DNA Rearrangements
- Large insertions or deletions - removing or adding entire exons or gene segments.
- Inversions - a segment is reversed in orientation within the chromosome.
- Translocations - a segment moves from one chromosome to another.
- These can be detected by Southern blotting (as noted in Lippincott, p. 1339).
6. Single Nucleotide Polymorphisms (SNPs - "snips")
- Lippincott states: ~90% of human genome variation comes in the form of SNPs, i.e., a variation involving just one base.
- They are the most common type of sequence variation at a given locus.
- Most occur in non-coding regions (introns/intergenic regions) and are harmless.
- Some SNPs in coding or regulatory regions can cause disease susceptibility or directly cause disease.
- The HapMap project catalogues common SNPs in the human genome.
b) The Most Common Error in DNA
Transitions are the most common type of point mutation, and among all DNA sequence variants:
Single Nucleotide Polymorphisms (SNPs) account for approximately 90% of all human genome variation - Lippincott Illustrated Reviews: Biochemistry, 8th ed., p. 1341.
More specifically:
- C → T transitions (particularly at CpG dinucleotide sites) are the single most frequent type of mutation in the human genome. This happens because cytosine residues at CpG sites are often methylated (forming 5-methylcytosine), and spontaneous deamination of 5-methylcytosine produces thymine - causing a C → T change. This is why CpG sites are mutation "hotspots."
Within the context of DNA replication errors:
- The most common replication errors are mismatched base pairs (e.g., G pairing with T instead of C), which if uncorrected produce transition mutations in the next round of replication.
- The proofreading activity of DNA polymerase III (3'→5' exonuclease) corrects most of these, and the mismatch repair (MMR) system catches the rest - making the overall fidelity of replication extremely high (~1 error per 10⁹ base pairs).
Summary Table
| Mutation Type | Description | Most Common? |
|---|---|---|
| SNPs (mostly transitions) | Single base changes | Yes - ~90% of all genome variation |
| Transitions (purine↔purine, pyrimidine↔pyrimidine) | Most common type of base substitution | Most common point mutation |
| Transversions (purine↔pyrimidine) | Less structurally compatible, less frequent | Less common |
| Frameshift (indels) | Reading frame shifted | Less common |
| Trinucleotide repeats | Repeat expansion | Uncommon |
| Large rearrangements | Inversions, translocations | Least common |
Source: Biochemistry, 8th ed. Lippincott Illustrated Reviews, Chapter 30 (DNA Repair), Chapter 34 (Biotechnology) - pp. 1339-1341.
Dna repair mechanism with clinical examples
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have complete, detailed content from Lippincott on all DNA repair mechanisms. Let me compile the comprehensive answer.
DNA Repair Mechanisms - Lippincott Illustrated Reviews: Biochemistry, 8th ed. (Chapter 30, pp. 1176-1187)
Overview
Despite the proofreading activity of DNA polymerase, errors can occur and DNA is constantly subjected to environmental insults - chemicals (e.g., nitrous acid, alkylating agents), radiation (UV, ionizing), and spontaneous base loss (~10,000 purine bases lost per cell per day). If damage is not repaired, a permanent mutation results, which can lead to cancer or cell death.
Most repair systems are excision repair systems that follow the same general 4-step strategy:
- Recognition of the lesion
- Excision (removal) of the damaged nucleotide(s)
- Gap filling using the undamaged complementary strand as template
- Ligation to restore strand continuity
Mechanism 1 - Mismatch Repair (MMR)
What it repairs: Replication errors (mismatched base pairs) that escape the proofreading activity of DNA pol III.
How it works:
- The Mut proteins (MutS, MutL, MutH in E. coli; MSH and MLH homologs in humans) identify the mispaired nucleotide(s).
- The system must distinguish the correct (parent) strand from the incorrect (daughter) strand.
- In prokaryotes: the parent strand is methylated at GATC sequences (by DAM methylase). The newly synthesized daughter strand is transiently unmethylated - this hemimethylation signals which strand needs correction.
- In eukaryotes: the daughter strand is identified by nicks in the newly synthesized strand.
- An endonuclease nicks the daughter strand; an exonuclease removes the mismatched nucleotide(s) and additional flanking bases.
- DNA pol III (prokaryotes) / pol δ (eukaryotes) fills the gap; DNA ligase seals it.
MMR reduces the error rate of replication from 1 in 10⁷ to 1 in 10⁹ nucleotides.
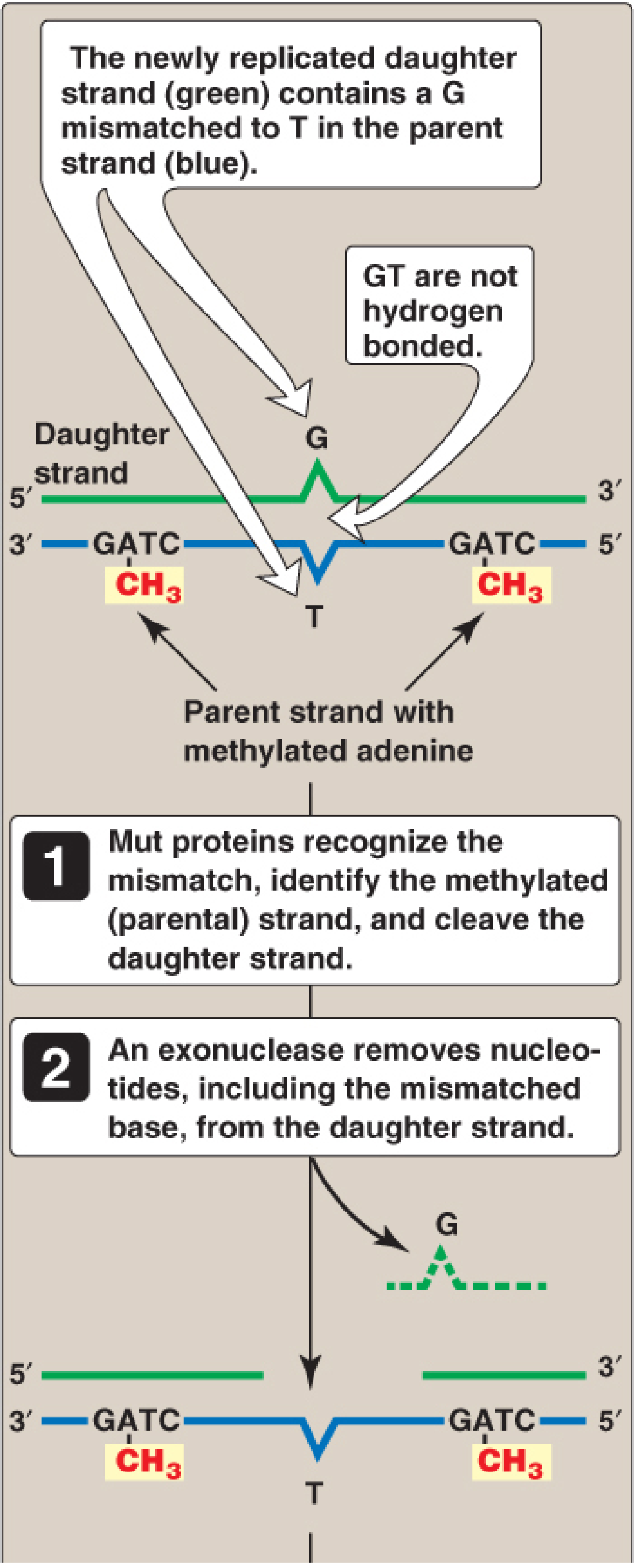
Clinical Example - Lynch Syndrome (HNPCC):
Defects in MMR proteins in humans cause Lynch syndrome (Hereditary Nonpolyposis Colorectal Cancer). Mutations in MSH2 and MLH1 (human homologs of Mut proteins) account for ~90% of Lynch syndrome cases. Although it confers strongly increased risk for colon cancer (and other cancers - endometrial, ovarian, gastric), only about 5% of all colon cancer results from MMR mutations.
Mechanism 2 - Nucleotide Excision Repair (NER)
What it repairs: Bulky DNA adducts - primarily pyrimidine (thymine) dimers caused by UV radiation. Also repairs chemical adducts such as guanine adducts from benzo[a]pyrene (cigarette smoke).
How it works:
- UV radiation causes covalent joining of two adjacent pyrimidines (usually T-T), forming an intrastrand cross-link (dimer) that blocks DNA replication.
- A UV-specific endonuclease (UvrABC excinuclease in bacteria) recognizes the bulky dimer.
- The enzyme cleaves the damaged strand on both the 5' and 3' sides of the lesion.
- A short oligonucleotide (~12 nucleotides in prokaryotes; ~27-29 nucleotides in eukaryotes) containing the dimer is excised.
- DNA pol I (prokaryotes) / pol δ or ε (eukaryotes) fills the gap.
- DNA ligase seals the nick.
- NER occurs throughout the cell cycle (not limited to S phase).
- NER has two sub-pathways: global genomic repair (searches all chromosomes) and transcription-coupled repair (preferentially repairs lesions encountered by RNA polymerase).
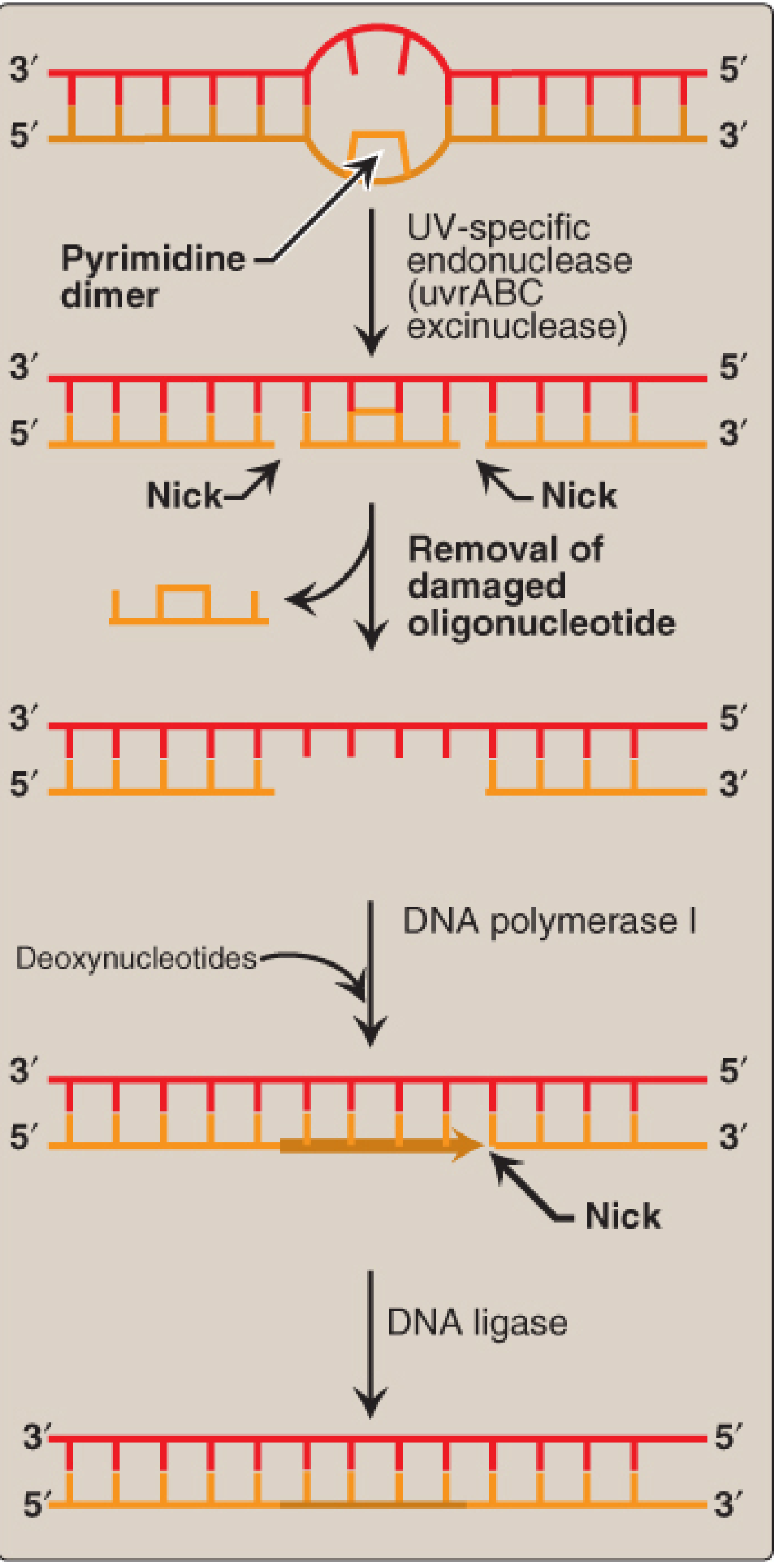
Clinical Example - Xeroderma Pigmentosum (XP):
In this rare autosomal recessive disease, skin cells cannot repair pyrimidine dimers caused by sunlight UV radiation. Unrepaired dimers accumulate, causing massive mutations, leading to early onset and numerous skin cancers (basal cell carcinoma, squamous cell carcinoma, melanoma). XP can be caused by defects in seven XP genes (XPA through XPG) that encode NER proteins.
Mechanism 3 - Base Excision Repair (BER)
What it repairs: Altered, damaged, or missing single bases - particularly:
- Uracil in DNA (from deamination of cytosine, or misincorporation of dUTP during replication)
- Oxidized bases (e.g., 8-oxoguanine)
- Alkylated bases (e.g., methylated adenine from dimethyl sulfate)
- AP sites (apurinic/apyrimidinic sites) - spontaneously lost bases (~10,000/cell/day are purines)
How it works:
- A specific DNA glycosylase recognizes the abnormal base and hydrolyzes the glycosidic bond, removing the base from the deoxyribose-phosphate backbone. This creates an AP site (apyrimidinic or apurinic site).
- A specific AP endonuclease cleaves the strand just to the 5' side of the AP site.
- A deoxyribose phosphate lyase removes the single base-free sugar-phosphate residue.
- DNA pol I (prokaryotes) fills the gap using the complementary strand.
- DNA ligase seals the nick.
Key point: BER removes one nucleotide at a time (single-nucleotide BER), unlike NER which removes an oligonucleotide.
Clinical Example - Spontaneous Mutation Hotspots:
Spontaneous deamination of cytosine → uracil in DNA is a major source of C → T mutations. If the uracil is not removed by BER (via uracil-DNA glycosylase) before the next replication cycle, a permanent C:G → T:A transition is created. This is why CpG sites are mutation hotspots in the human genome - they involve 5-methylcytosine, which deaminates to thymine (not uracil), and thymine is not recognized by uracil-DNA glycosylase.
Mechanism 4 - Double-Strand Break (DSB) Repair
What it repairs: Both strands broken simultaneously - the most dangerous type of DNA damage. Cannot be repaired by excision repair (no intact complementary strand available as template).
Caused by:
- Ionizing radiation (X-rays, gamma rays)
- Chemotherapeutic agents (e.g., doxorubicin, bleomycin)
- Oxidative free radicals
- Also occurs naturally during genetic recombination (meiosis)
Sub-mechanism A: Non-Homologous End Joining (NHEJ)
- Proteins recognize and bind the broken DNA ends.
- The ends are processed and ligated together directly.
- Error prone - some DNA is lost in the process (mutagenic).
- Can occur at any phase of the cell cycle.
- Defects in NHEJ are associated with cancer predisposition and immunodeficiency syndromes.
Sub-mechanism B: Homologous Recombination (HR)
- Uses the sister chromatid or homologous chromosome as a template to replace lost DNA.
- Much less error prone ("error free") because lost DNA is accurately replaced.
- Occurs in late S and G₂ phases of the cell cycle (when sister chromatid is available).
Clinical Examples:
- BRCA1 / BRCA2 mutations: BRCA1 and BRCA2 proteins are involved in homologous recombination. Mutations in these genes impair HR, leading to greatly increased risk for breast cancer and ovarian cancer. This is why BRCA-mutated cancers are sensitive to PARP inhibitors (which block a backup repair pathway).
- Ataxia-Telangiectasia (AT): Caused by mutations in the ATM gene (a kinase that senses DSBs and coordinates the DSB repair response). Clinical features include cerebellar ataxia, telangiectasias, immunodeficiency, and greatly increased cancer risk (especially lymphomas).
Summary Table
| Repair Mechanism | DNA Damage Repaired | Key Enzymes | Clinical Disease if Defective |
|---|---|---|---|
| Mismatch Repair (MMR) | Replication errors (mismatched bases) | Mut proteins (MSH2, MLH1 in humans) | Lynch Syndrome (HNPCC) |
| Nucleotide Excision Repair (NER) | Bulky adducts, pyrimidine dimers (UV) | UvrABC excinuclease; XP proteins | Xeroderma Pigmentosum (XP) |
| Base Excision Repair (BER) | Altered/missing single bases, AP sites | DNA glycosylase, AP endonuclease | MUTYH-associated polyposis |
| NHEJ (DSB repair) | Double-strand breaks | Ku proteins, DNA-PKcs, ligase IV | Immunodeficiency, cancer |
| Homologous Recombination (DSB repair) | Double-strand breaks | BRCA1, BRCA2, RAD51 | Breast/Ovarian cancer (BRCA mutations) |
Source: Biochemistry, 8th ed. Lippincott Illustrated Reviews, Chapter 30, pp. 1176-1187
This is a shared conversation. Sign in to Orris to start your own chat.