I’m a pathology final year resident help me prepare a 10 mark answer on imported infectious diseases
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Searching the Web
imported infectious diseases pathology exam answer India USMLE tropical parasitic diseases
I now have enough context to write a comprehensive, exam-grade answer. Let me compile this from expert pathology knowledge (Robbins, Muir's, and standard references).
Imported Infectious Diseases - 10 Mark Pathology Answer
Definition
Imported infectious diseases are infections acquired in one geographic region (usually tropical/subtropical) and diagnosed in another country, typically after international travel, immigration, or population movement. They represent a growing public health concern due to globalization, mass tourism, and climate change expanding vector habitats.
Classification
Imported infectious diseases are broadly classified as:
| Category | Examples |
|---|---|
| Protozoal | Malaria, Leishmaniasis, Trypanosomiasis, Amoebiasis, Giardiasis |
| Helminthic | Schistosomiasis, Filariasis, Strongyloidiasis, Cysticercosis, Hydatid disease |
| Viral | Dengue, Chikungunya, Viral Haemorrhagic Fevers (Ebola, Lassa, Marburg), Zika |
| Bacterial | Typhoid, Brucellosis, Cholera, Plague, Melioidosis |
| Mycobacterial | Tuberculosis, Leprosy |
1. Malaria
Organism: Plasmodium falciparum (most lethal), P. vivax, P. ovale, P. malariae, P. knowlesi
Vector: Female Anopheles mosquito
Pathological Features
Blood:
- Parasitized red cells - ring forms, trophozoites, schizonts, gametocytes on peripheral smear
- P. falciparum - Maurer's clefts; P. vivax - Schuffner's dots; P. malariae - band forms (trophozoites)
- Haemolytic anaemia - intravascular haemolysis releases haemozoin (malarial pigment)
Spleen:
- Acute: congestion, enlargement, soft consistency ("ague cake")
- Chronic: massive splenomegaly, grey-black pigmentation from haemozoin, fibrosis, hyperplastic RE cells
- Tropical splenomegaly syndrome - exaggerated immunological response
Liver:
- Hepatomegaly with haemozoin in Kupffer cells
- Centrizonal necrosis in severe falciparum malaria
- Periportal inflammation, haemosiderin deposition
Brain (Cerebral malaria - P. falciparum):
- Cytoadherence - parasitized RBCs adhere to cerebrovascular endothelium via PfEMP1 ligand and ICAM-1 receptors
- Duffy rosettes, sequestration in cerebral capillaries and venules
- Ring haemorrhages - Dürck's granulomas (necrotic foci surrounded by microglia)
- Petechial haemorrhages throughout white matter
- Brain oedema, raised intracranial pressure
Kidney:
- Blackwater fever (P. falciparum) - massive intravascular haemolysis - haemoglobinuria - acute tubular necrosis
- Quartan malarial nephropathy (P. malariae) - immune complex deposition - membranoproliferative glomerulonephritis
2. Leishmaniasis
Organism: Leishmania donovani (visceral), L. tropica/major (cutaneous), L. braziliensis (mucocutaneous)
Vector: Female Phlebotomus sandfly
Form: Amastigotes (Leishman-Donovan bodies) intracellularly in macrophages
Pathological Features
Visceral (Kala-azar - L. donovani):
- Massively enlarged spleen (can reach 3-4 kg) - sinusoidal dilatation, RE hyperplasia packed with amastigotes
- Liver - Kupffer cell hyperplasia laden with LD bodies; periportal fibrosis in chronic cases
- Bone marrow - hypercellular; macrophages with amastigotes, plasma cell infiltration
- Lymph nodes - cortical hyperplasia, sinus histiocytosis
- Skin - post-kala-azar dermal leishmaniasis (PKDL) - depigmented macules/nodules after treatment
- Profound anaemia, leukopenia, thrombocytopenia (pancytopenia due to hypersplenism + BM infiltration)
- Serology: hypergammaglobulinaemia, low albumin (reversed A:G ratio)
Cutaneous:
- Ulcerated skin lesion ("Delhi boil/Oriental sore") - granulomatous inflammation, giant cells, amastigotes in dermal macrophages, central necrosis
3. Schistosomiasis (Bilharziasis)
Organisms: S. mansoni (portal hypertension), S. haematobium (bladder), S. japonicum (portal hypertension, most severe)
Vector: Freshwater snail (Biomphalaria, Bulinus, Oncomelania)
Pathological Features
Acute (Katayama fever): immune complex-mediated - serum sickness-like reaction
Chronic - Hepatic (S. mansoni/japonicum):
- Pipestem/Symmer's clay pipestem fibrosis - periportal fibrosis around portal tracts due to granulomatous reaction to ova
- Granulomas with eosinophils, giant cells around ova - later fibrosis
- Portal hypertension - splenomegaly, oesophageal varices - preserved hepatocellular function (unlike cirrhosis)
- Liver surface: "hobnail" appearance; cut surface: white pipestem portal tracts
Bladder (S. haematobium):
- Sandy patches - calcified dead ova in bladder wall
- Squamous cell carcinoma of bladder - major complication (strong association)
- Obstructive uropathy - hydroureter, hydronephrosis
4. Viral Haemorrhagic Fevers (VHFs)
Examples: Ebola (Filoviridae), Lassa (Arenaviridae), Marburg (Filoviridae), Yellow Fever (Flaviviridae), Dengue
Pathological Features
General mechanism: Viral tropism for monocytes/macrophages - cytokine storm - endothelial injury - vascular permeability - haemorrhage - multi-organ failure
Liver:
- Ebola/Marburg/Yellow fever: councilman/acidophil bodies (eosinophilic apoptotic hepatocytes), mid-zonal necrosis (Yellow fever - zones 2), pan-lobular necrosis (Ebola)
- Viral inclusions (Ebola - intracytoplasmic)
Spleen: Lymphoid depletion, necrosis of periarteriolar sheaths
Kidneys: Acute tubular necrosis, glomerular changes
Dengue:
- Thrombocytopenia due to immune-mediated platelet destruction and BM suppression
- Dengue haemorrhagic fever (DHF) - plasma leakage, dengue shock syndrome
- Liver: hepatocyte apoptosis (Councilman-like bodies), Kupffer cell hypertrophy
5. Filariasis
Organisms: Wuchereria bancrofti, Brugia malayi/timori; Loa loa; Onchocerca volvulus
Vector: Mosquito (Culex, Anopheles, Aedes, Mansonia)
Pathological Features (Lymphatic Filariasis)
- Acute: lymphangitis, lymphadenitis with adult worms; eosinophilic infiltration, granuloma formation
- Chronic: obstruction of lymphatics - lymphoedema - elephantiasis (lower limbs, scrotum)
- Histology: filarial granuloma - central degenerated worm, palisading histiocytes, eosinophils, plasma cells, fibrosis
- Tropical pulmonary eosinophilia - microfilariae in lungs - interstitial pneumonitis, eosinophilia
Onchocerciasis ("River blindness"):
- Subcutaneous nodules (onchocercoma) - adult worms in fibrous capsule
- Ocular pathology: sclerosing keratitis, retinal atrophy due to microfilariae
6. Trypanosomiasis
African (Sleeping Sickness - T. brucei gambiense/rhodesiense):
- Chancre at bite site - perivascular lymphocytic infiltration
- Lymphadenopathy - Winterbottom's sign (posterior cervical)
- Encephalitis - demyelination, perivascular cuffing, Mott cells (plasma cells with Russell bodies) in brain
American (Chagas Disease - T. cruzi):
- Acute: pseudocysts in myocardium, skeletal muscle, neurons; nests of amastigotes
- Chronic: dilated cardiomyopathy - apical aneurysm (characteristic), conduction system fibrosis
- Megaoesophagus, megacolon - destruction of Auerbach's plexus (myenteric neurons)
Diagnostic Approach in Pathology
| Technique | Application |
|---|---|
| Thick/thin blood smear (Giemsa) | Malaria species identification |
| LD body (Giemsa stain) | Leishmaniasis in BM/splenic aspirate |
| Ziehl-Neelsen | Acid-fast organisms |
| Haematoxylin-eosin | Granulomas, tissue reaction |
| PAS / silver stains | Fungal infections |
| PCR / immunohistochemistry | VHFs, confirmation |
| Electron microscopy | Viral ultrastructure |
Summary Table
| Disease | Organism | Key Pathological Lesion | Classic Finding |
|---|---|---|---|
| Malaria | Plasmodium spp. | Haemolysis, haemozoin | Dürck's granulomas (cerebral) |
| Kala-azar | L. donovani | Splenomegaly, LD bodies | Reversed A:G ratio |
| Schistosomiasis | Schistosoma spp. | Pipestem fibrosis | Bladder SCC |
| Ebola/Yellow Fever | Filovirus/Flavivirus | Liver necrosis | Councilman bodies |
| Filariasis | Wuchereria bancrofti | Lymphatic obstruction | Elephantiasis |
| Chagas | T. cruzi | Dilated cardiomyopathy | Apical aneurysm, megacolon |
Examiner's Note - How to Score Full Marks
For a 10-mark answer in pathology finals, the examiner looks for:
- Definition and classification with examples (1.5 marks)
- Detailed pathology of at least 3-4 diseases (5 marks) - emphasis on gross and histological features
- Pathogenesis/mechanism briefly stated (1.5 marks)
- Diagnostic approach (1 mark)
- Complications mentioned (1 mark)
Prioritise: Malaria (cerebral malaria/blackwater fever), Leishmaniasis (LD bodies), and Schistosomiasis (pipestem fibrosis/SCC bladder) as these are highest-yield in most Indian and international pathology final exams. VHFs and Chagas are important if examiners have a global emphasis.
Sources: Robbins & Cotran Pathologic Basis of Disease 10e; Muir's Textbook of Pathology; Mandell's Principles and Practice of Infectious Diseases
PNH, 10 marker
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a complete, textbook-referenced 10-mark answer. Here it is:
Paroxysmal Nocturnal Haemoglobinuria (PNH) - 10 Mark Answer
Definition
Paroxysmal Nocturnal Haemoglobinuria (PNH) is an acquired clonal haematopoietic stem cell disorder characterised by the production of abnormal erythrocytes, granulocytes, and platelets that are deficient in GPI-anchored complement regulatory proteins, rendering them exquisitely susceptible to complement-mediated intravascular haemolysis.
It is the only haemolytic anaemia caused by an acquired genetic defect (somatic mutation).
Molecular Pathogenesis - The PIGA Mutation
The Gene
- PNH results from somatic mutations in the PIGA gene (Phosphatidylinositol Glycan complementation class A)
- PIGA is located on the X chromosome and is subject to lyonisation (random X inactivation)
- Because PIGA is X-linked, a single somatic mutation in the active PIGA allele of any haematopoietic stem cell is sufficient to produce a deficiency state
- All clonal progeny - red cells, white cells, and platelets - are deficient in GPI-linked proteins
-
120 distinct somatic PIGA mutations have been described
The GPI Anchor
Proteins are anchored to cell membranes in two ways:
- Transmembrane/integral - via hydrophobic span
- GPI-linked - via covalent linkage to glycosylphosphatidylinositol (GPI) lipid anchor
PIGA encodes an enzyme essential for GPI anchor synthesis. Its deficiency causes secondary loss of all GPI-anchored proteins from the cell surface.
Key GPI-Anchored Proteins Lost in PNH
| Protein | Alias | Function |
|---|---|---|
| CD59 | MIRL (Membrane Inhibitor of Reactive Lysis) | Inhibits C3 convertase; blocks MAC (C5b-9) assembly - most important |
| CD55 | DAF (Decay Accelerating Factor) | Antagonises C3/C5 convertase complexes |
| C8-binding protein | Homologous restriction factor | Controls MAC assembly |
| CD58 | LFA-3 | Cell adhesion |
| CD14 | Endotoxin receptor | Innate immunity |
| CD16a | FcγRIII | NK cell/neutrophil function |
| Acetylcholinesterase | AChE | Enzymatic (on RBCs) |
Why PNH Clones Expand - The Autoimmune Hypothesis
Normal individuals harbor small numbers of PIGA-mutant bone marrow cells but do not develop PNH. Clinically evident PNH arises only when these mutant cells have a selective survival advantage - hypothesised to occur when autoimmune attack (T-cell mediated) targets GPI-anchored antigens on normal stem cells, sparing GPI-deficient (PIGA-mutant) cells.
This elegantly explains the strong clinical association between PNH and aplastic anaemia - both share an autoimmune aetiology, and 50-60% of aplastic anaemia patients harbor subclinical PNH clones.
Three Types of PNH Red Cells (Henry's Classification)
| Type | Complement Sensitivity | GPI Proteins |
|---|---|---|
| Type I | Normal | Normal |
| Type II | 3-5x normal | Partial deficiency |
| Type III | 15-25x normal | Complete absence |
The proportion of Type III cells determines clinical severity.
Clinical Features and Pathology
1. Haemolytic Anaemia (Intravascular)
- Loss of CD59 allows uncontrolled activation of the alternative complement pathway on the RBC surface
- The C5b-9 membrane attack complex (MAC) forms and lyses RBCs intravascularly
- Classically paroxysmal and nocturnal - sleep causes mild CO₂ retention, slight fall in blood pH, increased complement activity
- Importantly: paroxysmal nocturnal haemoglobinuria in the classic sense occurs in only 25% of cases - chronic haemolysis without overt haemoglobinuria is more typical
- Bouts can be triggered by: infection, surgery, blood transfusion, contrast dyes, exercise
Lab findings:
- Normocytic/normochromic anaemia with reticulocytosis (often less than expected)
- Hypochromic microcytic change due to chronic iron loss in urine
- Elevated LDH, unconjugated bilirubin, reduced/absent haptoglobin
- Haemoglobinuria (pink/brown urine) - intravascular haemolysis
- Haemosiderinuria (almost constantly present) - Prussian blue stain on urine sediment
- Direct Coombs/DAT: negative (distinguishes from autoimmune haemolytic anaemia)
- Pancytopenia in many patients: neutropenia in 60%, thrombocytopenia in 66%
2. Thrombosis - The Major Cause of Death
- Thrombosis occurs in approximately 40% of PNH patients and is the leading cause of disease-related death
- 85% are venous thromboses, often at unusual sites:
- Hepatic vein - Budd-Chiari syndrome
- Portal vein
- Cerebral venous sinuses
- Abdominal veins
- Mesenteric veins
Mechanisms of thrombosis:
- Loss of CD59 on platelets → phosphatidylserine externalisation → platelet prothrombinase complex assembly → enhanced thrombin generation
- Free haemoglobin released during haemolysis scavenges nitric oxide (NO) → loss of NO-mediated vasodilation and platelet inhibition → endothelial injury, platelet activation
- MAC-mediated endothelial damage
Abdominal pain in ~33% of patients - attributed to mesenteric vein microthrombi and smooth muscle dystonia from NO scavenging by free Hb.
3. Bone Marrow Failure
- Marrow may be hypercellular (erythroid hyperplasia) or hypocellular
- PNH, aplastic anaemia, and myelodysplastic syndrome form a clinical continuum
- ~5% of PNH patients transform to AML or MDS over time
PNH Clinical Categories
- Classic PNH - haemolysis and thrombosis dominant, usually >50% PNH clone
- PNH in the setting of another BM disorder (aplastic anaemia, MDS) - cytopenias predominate
- Subclinical PNH - small clone (<1%), no haemolysis, found incidentally
Laboratory Diagnosis
Old Tests (Historical)
- Ham's test (Acidified serum lysis test): Patient's RBCs lysed in acidified normal serum (activates complement) - positive in PNH, negative in normal. Largely abandoned.
- Sucrose haemolysis test (Sugar water test): Low ionic strength sucrose activates complement - less specific. Also abandoned.
Current Gold Standard: Flow Cytometry
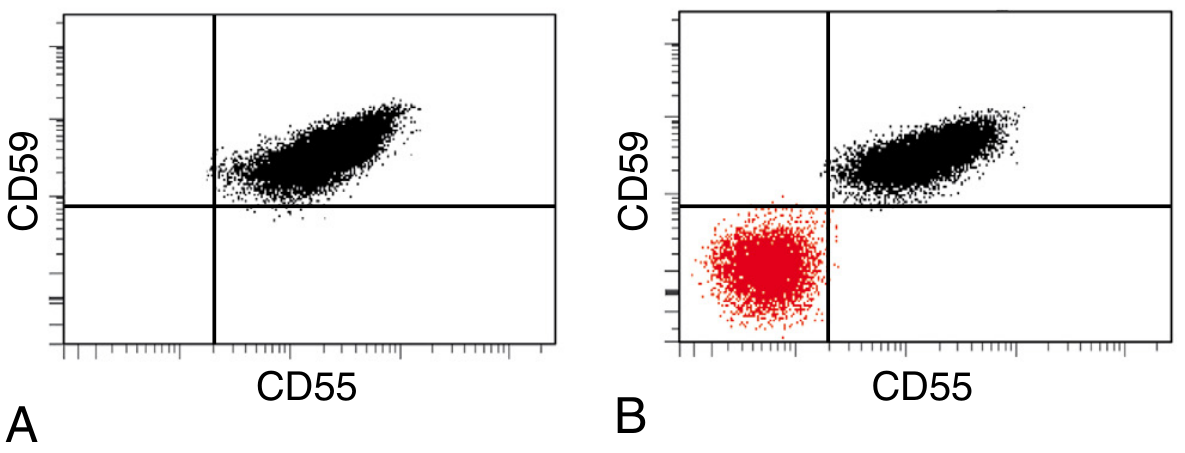
Fig. 14.13 (Robbins PBD 10e) - Flow cytometry in PNH. (A) Normal individual - all RBCs express CD55 and CD59. (B) PNH patient - a large CD55-/CD59- population (red) coexists with normal RBCs.
- Red cells: stained with anti-CD59, anti-CD55 monoclonal antibodies
- Granulocytes: anti-CD24 (excellent GPI-linked marker)
- Monocytes: anti-CD14 (GPI-linked)
- FLAER (Fluorescein-labelled aerolysin): binds directly to GPI anchor - increasingly used as gold standard, highly sensitive for detecting small clones
- High-sensitivity flow cytometry can detect clones as small as 0.01%
Additional labs
- LDH (elevated - marker of intravascular haemolysis)
- Urine haemosiderin (Prussian blue stain - almost always positive)
- Peripheral smear: normocytic anaemia, variable reticulocytosis
- No schistocytes (distinguishes from TMA/TTP)
Treatment
| Modality | Details |
|---|---|
| Eculizumab | Humanised anti-C5 monoclonal antibody; blocks MAC formation; approved 2007. Reduces haemolysis, transfusion requirements, thrombosis risk by up to 90%. Requires IV dosing every 14 days. Risk: meningococcal infection (meningococcal vaccination mandatory before use) |
| Ravulizumab | Long-acting anti-C5 antibody; dosing every 8 weeks |
| Pegcetacoplan | Anti-C3 (proximal complement inhibitor); prevents both intravascular AND extravascular haemolysis; corrects anaemia more completely |
| HSCT | Only curative option; reserved for intercurrent aplastic anaemia, myeloid malignancy, or refractory disease |
| Supportive | Iron/folate replacement, anticoagulation for thrombosis, transfusions |
| Immunosuppression | Useful when marrow aplasia is prominent (cyclosporine, ATG) |
Important note on eculizumab: By blocking MAC at C5, it protects PNH RBCs from intravascular lysis. However, PNH cells still bind C3b fragments and become opsonised → iatrogenic extravascular haemolysis in macrophages; DAT becomes positive. Pegcetacoplan (C3 blockade) prevents this.
Summary - Key Facts for Exam
| Feature | Detail |
|---|---|
| Gene | PIGA (X-linked, somatic mutation) |
| Anchor lost | GPI (glycosylphosphatidylinositol) |
| Most important protein lost | CD59 (MIRL) |
| Type of haemolysis | Intravascular (complement-mediated) |
| Classic presentation | Morning haemoglobinuria, anaemia, venous thrombosis |
| Diagnostic test | Flow cytometry (FLAER/CD59/CD55) |
| Old tests | Ham's acid lysis, sucrose haemolysis (obsolete) |
| Thrombosis sites | Hepatic vein (Budd-Chiari), portal, cerebral |
| Coombs/DAT | Negative (unlike autoimmune HA) |
| Leading cause of death | Thrombosis |
| Transformation | 5% → AML/MDS |
| Treatment | Eculizumab (anti-C5); HSCT for cure |
Examiner's Scoring Guide
For full 10 marks, cover:
- Definition and unique nature (acquired, only haemolytic anaemia with acquired genetic defect) - 1 mark
- PIGA gene, X-linkage, somatic mutation mechanism - 2 marks
- GPI anchor biology and which proteins are lost (CD55, CD59, C8BP) - 1.5 marks
- Pathogenesis of haemolysis, thrombosis, pancytopenia - 2 marks
- Clinical types of RBCs (I/II/III) and clinical features - 1 mark
- Lab diagnosis - flow cytometry, FLAER, Ham's test (historical) - 1.5 marks
- Treatment - eculizumab mechanism, HSCT - 1 mark
Sources: Robbins & Cotran Pathologic Basis of Disease 10e (p. 606); Henry's Clinical Diagnosis and Management by Laboratory Methods; Harrison's Principles of Internal Medicine 22e
Classify myelodysplastic syndrome And mention diagnostic criteria for 10 marks
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I have rich content from both Robbins PBD 10e and Henry's. The textbook content covers pathogenesis, morphology, cytogenetics, and clinical features thoroughly. Let me now compile the full 10-mark answer using this plus my expert knowledge of the WHO 2022 / ICC 2022 classification - the current standard for MDS.
Myelodysplastic Neoplasms (MDS) - Classification and Diagnostic Criteria - 10 Mark Answer
Definition
Myelodysplastic Neoplasms (MDS) - formerly called Myelodysplastic Syndromes - are a group of clonal haematopoietic stem cell disorders characterised by:
- Maturation defects and ineffective haematopoiesis
- Peripheral blood cytopenias despite a normo- or hypercellular marrow
- Morphological dysplasia in one or more myeloid cell lines
- Genetic instability with a high risk of transformation to AML (10-40% of cases)
The term "Myelodysplastic Neoplasm" is preferred in the WHO 5th edition (2022) and ICC 2022 classification, reflecting their established neoplastic nature, though "MDS" remains in widespread clinical use.
Aetiology and Types
| Type | Details |
|---|---|
| Primary (de novo) MDS | Idiopathic; no prior genotoxic exposure |
| Secondary / therapy-related MDS (t-MDS) | Follows chemotherapy (alkylating agents, topoisomerase II inhibitors) or radiation therapy; appears 2-8 years after exposure; more aggressive, rapid AML transformation |
Precursor state: Clonal Haematopoiesis of Indeterminate Potential (CHIP) - normal blood counts + clonal driver mutation (same mutations as MDS); progresses to MDS at ~1% per year.
Pathogenesis
Driver mutations group into three major functional categories (Robbins PBD 10e):
1. Epigenetic Regulators
- DNA methylation: TET2, DNMT3A, IDH1/IDH2
- Histone modification: EZH2, ASXL1
- Dysregulation of the epigenome - transcriptional silencing of differentiation genes
2. RNA Splicing Factors (characteristic of MDS)
- SF3B1 - strongly associated with ring sideroblasts; relatively favourable prognosis
- SRSF2, U2AF1, ZRSR2
- Alter mRNA processing affecting tumour suppressors and oncogenes
- Patients with CHIP + splicing factor mutations are at particularly high risk for MDS
3. Transcription Factors
- RUNX1, ETV6 - loss-of-function mutations impairing normal myelopoiesis
- Note: Common AML translocations [t(8;21), inv(16), t(15;17)] are absent in MDS
TP53 Mutations
- ~10% of MDS cases; correlate with complex karyotype and worst prognosis; biallelic TP53 mutations define a distinct entity
Recurrent Chromosomal Abnormalities
Predominantly loss of genetic material (unlike AML which has balanced translocations):
- Monosomy 5 / del(5q) - loss of RPS14 → ineffective erythropoiesis
- Monosomy 7 / del(7q)
- del(20q), del(12p), del(11q), del(9q)
- Trisomy 8 (gain of MYC - stimulates cell growth)
- Isochromosome 17q
Morphological Features (Dysplasia)
Dysplasia is defined as morphological abnormalities in ≥10% of cells in a given lineage.
Erythroid Dysplasia
- Megaloblastoid change (nuclear-cytoplasmic asynchrony)
- Ring sideroblasts - iron deposits in mitochondria around nucleus (≥15% of erythroid precursors, or ≥5% if SF3B1 mutation present)
- Nuclear budding, multinucleation, karyorrhexis
- Cytoplasmic vacuolation
Granulocyte Dysplasia
- Pseudo-Pelger-Huet anomaly - bilobed or hyposegmented neutrophils (most characteristic)
- Hypogranular neutrophils (agranular cytoplasm)
- Nuclear hypo- or hypersegmentation
- Abnormal chromatin clumping
Megakaryocyte Dysplasia
- Micromegakaryocytes (small size, single nucleus) - most specific for MDS
- Large megakaryocytes with multiple separate nuclei (separated nuclear lobes)
- Hypolobated/monolobated forms
Classification
WHO 5th Edition (2022) and ICC (International Consensus Classification) 2022
Both classifications represent a major shift - from morphology-based to morphology + genetics integrated classification.
WHO 5th Edition (2022) MDS Categories
| WHO 2022 Category | Key Features | Blast % (BM) |
|---|---|---|
| MDS with low blasts (MDS-LB) | 1-2 lineage dysplasia, cytopenias, no ring sideroblasts | <5% BM, <2% PB |
| MDS with low blasts and SF3B1 mutation (MDS-SF3B1) | Ring sideroblasts ≥15% (or ≥5% + SF3B1 mutation); typically unilineage erythroid dysplasia | <5% BM, <2% PB |
| MDS with low blasts and 5q deletion (MDS-5q) | del(5q) ± one additional abnormality (not -7/del7q); isolated macro-ovalocytes; hypolobated megakaryocytes | <5% BM, <2% PB |
| MDS with increased blasts 1 (MDS-IB1) | 5-9% blasts in BM or 2-4% in PB | 5-9% BM or 2-4% PB |
| MDS with increased blasts 2 (MDS-IB2) | 10-19% blasts in BM or 5-19% in PB or Auer rods | 10-19% BM or 5-19% PB |
| MDS with biallelic TP53 inactivation (MDS-biTP53) | Two TP53 mutations or one mutation + del(17p)/LOH; complex karyotype; very poor prognosis | Any blast % |
| MDS, hypoplastic (MDS-h) | Hypocellular marrow (<25% age-adjusted cellularity) + dysplasia; overlap with aplastic anaemia | <5% BM |
| MDS, not otherwise specified (MDS-NOS) | Does not fit above categories | <5% BM |
Prior WHO 2016 Classification (Still Widely Tested in Exams)
| WHO 2016 Category | Dysplastic Lineages | Cytopenias | RS% | BM Blasts | PB Blasts | Cytogenetics |
|---|---|---|---|---|---|---|
| MDS-SLD (Single lineage dysplasia) | 1 | 1-2 | <15% / <5%* | <5% | <1% | Any except MDS-defining |
| MDS-MLD (Multilineage dysplasia) | 2-3 | 1-3 | <15% / <5%* | <5% | <1% | Any except MDS-defining |
| MDS-RS-SLD (Ring sideroblasts, single lineage) | 1 | 1-2 | ≥15% / ≥5%* | <5% | <1% | Any except MDS-defining |
| MDS-RS-MLD (Ring sideroblasts, multilineage) | 2-3 | 1-3 | ≥15% / ≥5%* | <5% | <1% | Any except MDS-defining |
| MDS with isolated del(5q) | 1-2 | 1-2 | None/any | <5% | <1% | del(5q) alone or + 1 abnormality (not -7) |
| MDS-EB-1 (Excess blasts 1) | 0-3 | 1-3 | None/any | 5-9% | 2-4% | Any |
| MDS-EB-2 (Excess blasts 2) | 0-3 | 1-3 | None/any | 10-19% | 5-19% or Auer rods | Any |
| MDS-U (Unclassifiable) | Various | 1-3 | <15% | <5% | <1% | MDS-defining abnormality |
*≥5% if SF3B1 mutation present (RS = ring sideroblasts)
Diagnostic Criteria
Minimum Requirements for MDS Diagnosis
Prerequisite conditions (all must be met):
- Cytopenia in ≥1 lineage:
- Hb <13 g/dL (male) / <12 g/dL (female)
- ANC <1.8 × 10⁹/L
- Platelets <150 × 10⁹/L
- Exclusion of reactive causes of dysplasia (vitamin B12/folate deficiency, copper deficiency, HIV, drugs, alcohol, heavy metal toxicity, hypothyroidism)
- Stable cytopenia for ≥4 months (unless MDS-defining cytogenetic abnormality present)
MDS-defining criteria (≥1 required):
| Criterion | Threshold |
|---|---|
| Dysplasia | ≥10% of cells in ≥1 myeloid lineage |
| Ring sideroblasts | ≥15% of erythroid precursors (or ≥5% + SF3B1 mutation) |
| Increased blasts | 5-19% in BM or 2-19% in PB |
| MDS-defining cytogenetic abnormality | See below - sufficient even without morphological dysplasia |
MDS-Defining Cytogenetic Abnormalities (Henry's / WHO)
These are presumptive evidence of MDS in the context of unexplained cytopenia, even without dysplasia:
Unbalanced (loss):
- Monosomy 7 or del(7q)
- del(5q)
- Isochromosome 17q or t(17p)
- Monosomy 13 or del(13q)
- del(11q), del(12p) or t(12p), del(9q)
- idic(X)(q13)
Balanced (translocations):
- t(11;16)(q23.3;p13.3)
- t(3;21)(q26.2;q22.1)
- t(1;3)(p36.3;q21.2)
- t(2;11)(p21;q23.3)
- inv(3)(q21.3q26.2) / t(3;3)(q21.3;q26.2)
- t(6;9)(p23;q34.1)
Note: Trisomy 8, del(20q), and loss of Y chromosome are NOT considered MDS-defining as isolated abnormalities - they require concurrent morphological dysplasia.
Diagnostic Work-up
| Investigation | Finding in MDS |
|---|---|
| Peripheral blood smear | Cytopenias, pseudo-Pelger-Huet cells, hypogranular neutrophils, macrocytes, teardrop cells |
| Bone marrow aspirate | Dysplasia in ≥1 lineage, blast %, ring sideroblasts (Perls' stain) |
| Bone marrow biopsy | Cellularity, reticulin fibrosis, blast distribution |
| Cytogenetics (karyotype) | Clonal chromosomal abnormalities - required in all new MDS |
| FISH | For specific abnormalities when karyotype inadequate (<20 metaphases) |
| Flow cytometry | Aberrant antigen expression on myeloid precursors (supportive) |
| NGS/molecular panel | SF3B1, SRSF2, TET2, DNMT3A, ASXL1, RUNX1, TP53 - aids diagnosis and prognosis |
| Serum B12, folate, copper, TSH | Exclusion of nutritional/reactive dysplasia |
| Iron studies | Iron deficiency vs. sideroblastic process |
Prognosis - IPSS-R Scoring
The Revised International Prognostic Scoring System (IPSS-R) assigns points based on:
| Parameter | Variables |
|---|---|
| Cytogenetics | Very good → Very poor (5 categories) |
| BM blast % | <2%, 2-4.9%, 5-10%, >10% |
| Haemoglobin | <8, 8-10, ≥10 g/dL |
| Platelets | <50, 50-100, ≥100 × 10⁹/L |
| ANC | <0.8, ≥0.8 × 10⁹/L |
Risk groups: Very Low, Low, Intermediate, High, Very High - determines median OS and AML transformation risk.
Treatment Overview
| Strategy | Indication |
|---|---|
| Supportive (transfusions, ESAs, G-CSF) | Low-risk MDS, elderly patients |
| Lenalidomide | MDS with del(5q) - targets haploinsufficiency of RPS14 and CSNK1A1 |
| DNA hypomethylating agents (Azacitidine, Decitabine) | Higher-risk MDS; "reprogram" epigenetically dysregulated stem cells |
| Allogeneic HSCT | Only curative option; reserved for higher-risk younger patients |
| Luspatercept | MDS-SF3B1 / ring sideroblast subtypes - reduces transfusion burden |
Summary Table - Key Distinguishing Features
| Entity | Blasts | Key Feature | Prognosis |
|---|---|---|---|
| MDS-LB / MDS-SLD | <5% | 1-lineage dysplasia | Relatively good |
| MDS-SF3B1 / RS subtypes | <5% | Ring sideroblasts + SF3B1 | Favourable |
| MDS-5q | <5% | del(5q), hypolobated megakaryocytes | Favourable (responds to lenalidomide) |
| MDS-IB1 / EB-1 | 5-9% | Excess blasts | Intermediate |
| MDS-IB2 / EB-2 | 10-19% | Excess blasts / Auer rods | Poor; near-AML |
| MDS-biTP53 | Any | Biallelic TP53, complex karyotype | Very poor |
Examiner's Scoring Guide
For 10 marks:
- Definition, primary vs. secondary, CHIP precursor - 1.5 marks
- Pathogenesis (3 mutation categories + cytogenetics) - 2 marks
- Morphological features / dysplasia in each lineage - 1.5 marks
- Classification (WHO 2016 OR WHO 2022 table with key entities) - 2.5 marks
- Diagnostic criteria (cytopenias, dysplasia threshold, cytogenetic abnormalities) - 1.5 marks
- Prognosis/IPSS-R mention + treatment overview - 1 mark
Sources: Robbins & Cotran Pathologic Basis of Disease 10e; Robbins & Kumar Basic Pathology 11e; Henry's Clinical Diagnosis and Management by Laboratory Methods; WHO Classification of Haematolymphoid Tumours 5th ed. (2022)
Hemophagocytic Syndromes for 10 marks
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have everything. Here is the comprehensive 10-mark answer:
Hemophagocytic Syndromes (Hemophagocytic Lymphohistiocytosis - HLH) - 10 Mark Answer
Definition
Hemophagocytic Lymphohistiocytosis (HLH) is a life-threatening syndrome of pathological immune activation characterised by uncontrolled systemic activation of CD8+ cytotoxic T lymphocytes and macrophages, resulting in:
- Widespread phagocytosis of blood cells and their precursors (haemophagocytosis)
- Severe cytopenias
- Hypercytokinaemia ("cytokine storm")
- Multi-organ failure
It is also called Haemophagocytic Syndrome (HPS) or, when arising in the context of rheumatological disease, Macrophage Activation Syndrome (MAS).
Classification
HLH is broadly divided into Primary (Familial) and Secondary (Acquired) forms:
A. Primary HLH (Familial / Genetic)
1. Familial HLH (FHL) - Most Common
Autosomal recessive; caused by mutations in genes encoding the cytotoxic granule exocytosis machinery of CTLs and NK cells:
| Subtype | Gene | Protein | Step Affected |
|---|---|---|---|
| FHL-2 | PRF1 | Perforin | Granule content - pore forming protein |
| FHL-3 | UNC13D | Munc13-4 | Granule docking |
| FHL-4 | STX11 | Syntaxin-11 | Granule priming |
| FHL-5 | STXBP2 | Munc18-2 | Granule fusion with plasma membrane |
PRF1 and UNC13D variants account for ~30% each of FHL cases.
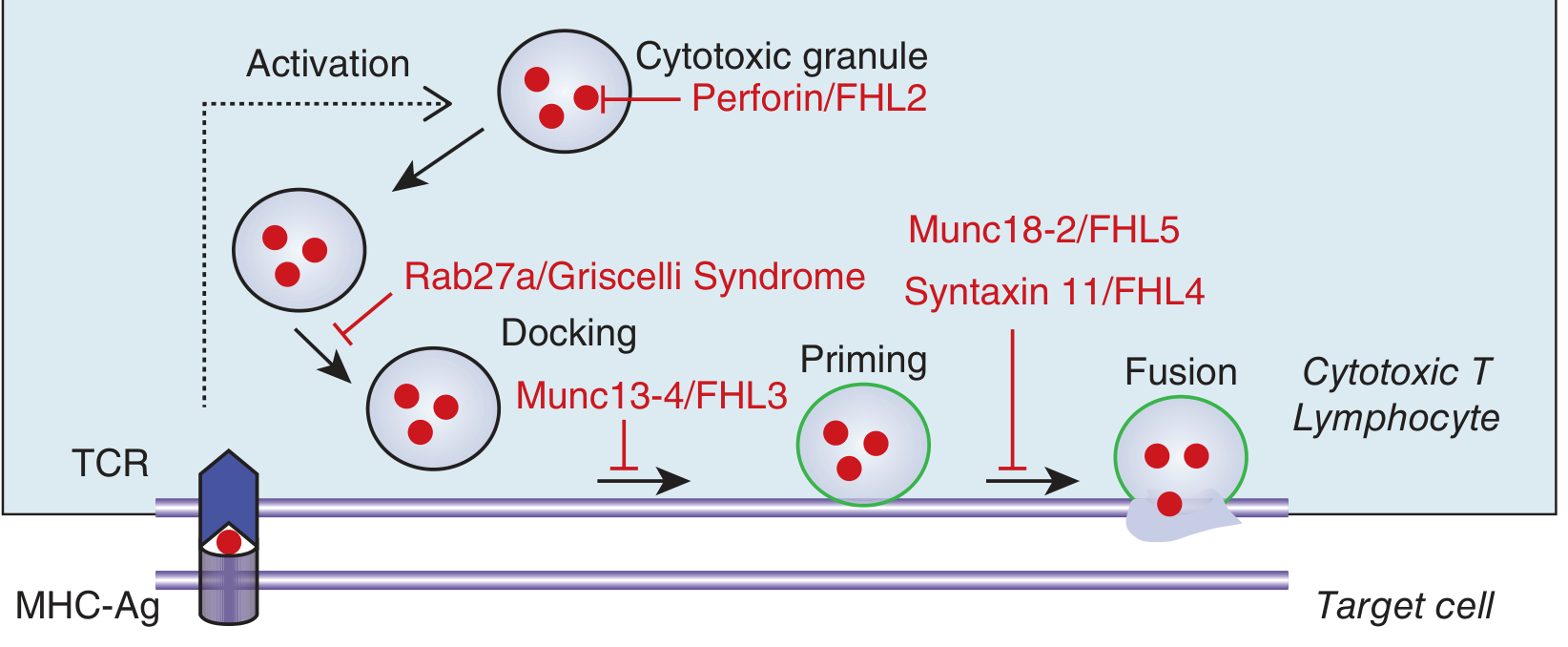
Fig. 68-1 (Harrison's 22e) - Genetic defects in FHL mapped to specific steps of cytotoxic granule exocytosis
2. Primary HLH with Partial Albinism
Three conditions combine HLH with abnormal pigmentation (hair examination aids diagnosis):
| Syndrome | Gene | Key Feature |
|---|---|---|
| Chédiak-Higashi syndrome | LYST | Giant lysosomes in leukocytes; progressive neurological disease; silver-grey hair |
| Griscelli syndrome type 2 | RAB27A | Hypopigmentation, silver-coloured hair; RAB27A impairs granule docking |
| Hermansky-Pudlak syndrome type II | AP3B1 | Albinism + platelet defect |
3. X-Linked Lymphoproliferative Disorder (XLP)
- XLP-1: SH2D1A gene → deficient SAP adaptor protein → triggered by EBV infection; impaired NK cell 2B4-mediated cytotoxicity; defective antigen-induced T-cell death
- XLP-2: BIRC4 gene → XIAP deficiency → HLH + colitis; linked to NLRC4 inflammasome dysregulation
4. Other Primary HLH
- Inflammasome activation disorders (NLRC4 gain-of-function)
- Inborn errors of immunity and metabolism
B. Secondary HLH (Acquired)
Triggered by an external stimulus in an individual with or without a pre-existing genetic predisposition:
| Trigger Category | Examples |
|---|---|
| Infectious (most common) | EBV (most frequent trigger overall), CMV, HSV, HIV, influenza, COVID-19, visceral leishmaniasis, tuberculosis |
| Malignancy-associated | Peripheral T-cell lymphoma (most common haematological trigger), NK/T-cell lymphoma, DLBCL, leukaemia |
| Autoimmune/Rheumatological (MAS) | Systemic JIA (most common), adult-onset Still's disease, SLE, Kawasaki disease |
| Iatrogenic | CAR-T cell therapy (cytokine release syndrome), checkpoint inhibitors |
| Idiopathic | No identifiable trigger |
Pathogenesis
The Central Defect
Under normal conditions: CTLs and NK cells detect and kill abnormal/infected cells via perforin-granzyme granule exocytosis - a sequence of granule activation → docking → priming → fusion with plasma membrane → perforin forms pores → granzymes enter target cell → apoptosis.
In FHL/primary HLH: this cytotoxic machinery is defective at one of several steps (as shown in the diagram above). The results are:
- Inability to kill infected/abnormal target cells - particularly virus-infected cells (e.g., EBV-infected B cells)
- Prolonged CTL-target cell engagement - the CTL remains locked in immunological synapse, secreting cytokines instead of lysing
- Massive IFN-γ secretion by overactivated CD8+ T cells and NK cells - the key proximal driver
- IFN-γ activates macrophages - producing unbridled macrophage activation
- Macrophage cytokine storm: TNF-α, IL-6, IL-12, IL-1β, IL-18, CXCL9 - systemic effects resembling sepsis (SIRS)
- Haemophagocytosis - activated macrophages engulf RBCs, platelets, WBCs and their precursors in BM, liver, spleen, and lymph nodes
- Vicious cycle: inflamed, non-dying target cells continue stimulating CTLs → further IFN-γ → further macrophage activation
In secondary HLH: heterozygous mutations in FHL genes lower the threshold for HLH, and an overwhelming antigenic stimulus (virus, tumour) drives the same cytokine storm even without complete loss of cytotoxic function.
Morphological Features
Bone Marrow
- Haemophagocytosis - macrophages engulfing intact RBCs, nucleated erythroid precursors, platelets, and granulocytes (the defining histological finding, though neither sensitive nor specific)
- May progress to hypocellularity in late disease
- Reactive lymphocytes and plasma cells
- Erythroid hyperplasia initially
Liver
- Periportal and sinusoidal lymphohistiocytic infiltrate
- Kupffer cell hyperplasia with haemophagocytosis
- Hepatocyte necrosis (resembles chronic persistent hepatitis on biopsy)
- Cholestasis
Spleen
- Expansion of red pulp by macrophages with haemophagocytosis
- Lymphoid depletion in severe/late disease
Lymph Nodes
- Sinus histiocytosis with haemophagocytosis
- Paracortical expansion by T cells and macrophages
CNS
- Perivascular lymphohistiocytic infiltrate
- Microglial activation
- Demyelination in severe cases
Clinical Features
Epidemiology
- Primary FHL: incidence 1 in 50,000 live births; median onset 3-6 months; autosomal recessive (equal M:F); more common in consanguineous populations
- Secondary HLH: any age; adults predominantly
Symptoms and Signs
| System | Features |
|---|---|
| Constitutional | High-grade fever (often prolonged, non-remitting) |
| Haematological | Pancytopenia (thrombocytopenia most prominent), petechia, purpura |
| Hepatosplenic | Hepatosplenomegaly, jaundice (conjugated hyperbilirubinaemia), elevated transaminases, LFT derangement |
| Lymphoid | Lymphadenopathy (~50%) |
| CNS | Seizures, altered consciousness, meningism, cranial nerve palsies, ataxia (~33% in FHL) |
| Skin | Transient maculopapular rash, oedema |
| GI | Diarrhoea (especially STXBP2 mutations) |
| Coagulation | DIC, severe bleeding, low fibrinogen |
Diagnostic Criteria - HLH-2004 (Updated 2024)
The diagnosis requires 5 of the following 7 criteria (as per 2024 Histiocyte Society update - NK cell activity criterion removed):
| Criterion | Threshold |
|---|---|
| 1. Fever | Temperature ≥38.5°C |
| 2. Splenomegaly | Clinical/imaging |
| 3. Bicytopenia | ≥2 cell lines: Hb <90 g/L (<100 g/L in infants <4 weeks), Platelets <100 × 10⁹/L, ANC <1.0 × 10⁹/L |
| 4. Hypertriglyceridaemia and/or hypofibrinogenaemia | Fasting TG ≥3.0 mmol/L (≥265 mg/dL) and/or Fibrinogen ≤1.5 g/L |
| 5. Haemophagocytosis in BM / spleen / lymph nodes | Histological |
| 6. Low or absent NK cell activity | Functional assay |
| 7. Ferritin ≥500 μg/L | (Very high levels >10,000 μg/L highly suggestive) |
| 8. Soluble CD25 (sIL-2Rα) ≥2400 U/mL | Marker of T-cell activation |
Note: The 2024 revision removed NK cell activity as a formal criterion (now 5/7 required). Molecular confirmation of a pathogenic FHL gene variant alone is sufficient for diagnosis, independent of clinical criteria.
HScore - Alternative Probabilistic Tool
The HScore calculates probability of HLH based on 9 parameters (temperature, organomegaly, cytopenias, ferritin, triglycerides, fibrinogen, transaminases, haemophagocytosis, known immunosuppression). Score >169 gives >93% probability of HLH. Particularly useful in secondary/adult HLH where criteria may not all be met.
Laboratory Work-up
| Test | Finding |
|---|---|
| CBC | Pancytopenia (thrombocytopenia earliest and most common) |
| Ferritin | Markedly elevated - >10,000 μg/L highly suggestive |
| sIL-2Rα (sCD25) | Elevated - marker of T-cell activation |
| Triglycerides | Elevated (hypertriglyceridaemia) |
| Fibrinogen | Reduced (hypofibrinogenaemia) |
| LFTs | Elevated transaminases, bilirubin |
| Coagulation | Prolonged PT/APTT, low fibrinogen, elevated D-dimer (DIC) |
| NK cell function | Low/absent NK cell cytotoxicity |
| Perforin expression | Flow cytometry for intracellular perforin in CTLs (absent in PRF1 mutations) |
| CD107a degranulation assay | Tests granule exocytosis - abnormal in UNC13D, STXBP2, STX11, RAB27A |
| BM biopsy/aspirate | Haemophagocytosis |
| Genetic sequencing (NGS) | Confirms FHL gene variants (PRF1, UNC13D, STX11, STXBP2, RAB27A, SH2D1A) |
| EBV/CMV/HIV serology | Identify infectious triggers |
Treatment
HLH-94 / HLH-2004 Protocol (Primary HLH)
Induction (weeks 1-8):
- Dexamethasone (penetrates blood-brain barrier) - suppresses inflammatory cytokines
- Etoposide - potent selective depletion of activated T cells; suppresses cytokine production
- Cyclosporin A - reduces T-cell cytokine production; added in HLH-2004
CNS-directed therapy: Intrathecal methotrexate for CNS disease
Continuation (weeks 9-40): Pulses of dexamethasone + etoposide + cyclosporin A as bridge to transplant
Definitive cure: Allogeneic HSCT - replaces the defective immune system; recommended for all primary HLH cases and for relapsed/refractory secondary HLH
Novel Targeted Therapies
- Emapalumab (anti-IFN-γ monoclonal antibody) - FDA approved; blocks the central cytokine driver in primary HLH
- Ruxolitinib (JAK1/2 inhibitor) - blocks JAK-STAT signalling downstream of IFN-γ; used especially in secondary/MAS-HLH
- Anakinra (IL-1 receptor antagonist) - used in MAS/sJIA-associated HLH
Secondary HLH
- Treat the underlying trigger - antimicrobials for infection, chemotherapy for malignancy
- MAS: High-dose corticosteroids ± cyclosporin, anakinra; cytotoxic therapy less preferred (risk of worsening the underlying rheumatological disease)
Prognosis
| Form | Prognosis |
|---|---|
| FHL untreated | Median survival 1-2 months; rapidly fatal |
| FHL treated (with HSCT) | ~50% long-term survival; significant neurological sequelae in ~33% |
| Secondary HLH | Depends on trigger and speed of treatment; malignancy-associated has worst prognosis |
| Long-term complications | Psychomotor retardation (25%), epilepsy (10%), ADHD (10%), endocrinopathies (~10%) |
Summary Table
| Feature | Primary HLH | Secondary HLH / MAS |
|---|---|---|
| Age | Infants, children | Any age |
| Genetics | Autosomal recessive / X-linked | Usually no germline mutation |
| Trigger | Viral (especially EBV) | Infection, malignancy, autoimmune |
| NK/CTL function | Absent/severely reduced | Variably reduced |
| Treatment | HLH-2004 protocol + HSCT | Treat trigger + corticosteroids ± etoposide |
| Cure | HSCT only | Treat underlying cause |
Examiner's Scoring Guide
For full 10 marks:
- Definition + classification (Primary/Secondary with subtypes) - 2 marks
- Pathogenesis - perforin-granzyme machinery, IFN-γ, cytokine storm - 2.5 marks
- Morphological features (BM, liver, spleen) - 1 mark
- Clinical features - 1 mark
- HLH-2004 diagnostic criteria (all 8 listed with thresholds) - 2 marks
- Treatment - HLH-2004 protocol, HSCT, emapalumab - 1.5 marks
Sources: Robbins & Cotran Pathologic Basis of Disease 10e (p. 581); Robbins & Kumar Basic Pathology 11e; Harrison's Principles of Internal Medicine 22e (Chapter 68, 2025)
Anemia of chronic diseases
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
I have all the textbook content needed. Now I'll compile the complete 10-mark answer.
Anaemia of Chronic Disease (ACD) / Anaemia of Inflammation (AI) - 10 Mark Answer
Definition
Anaemia of Chronic Disease (ACD), now more precisely termed Anaemia of Inflammation (AI), is the second most common cause of anaemia worldwide (after iron deficiency anaemia). It is characterised by:
- Mild to moderate anaemia in the setting of chronic infection, inflammation, or malignancy
- Low serum iron despite adequate or increased body iron stores
- Reduced erythropoietin (EPO) response
- Blunted bone marrow erythroid response
- The central mechanism is hepcidin-mediated functional iron deficiency - iron is trapped in reticuloendothelial macrophages and unavailable for erythropoiesis
Up to 40% of all anaemias worldwide have a significant ACD component. It is particularly prevalent in elderly patients.
Causes / Associated Conditions
ACD occurs in any disorder characterised by sustained immune activation:
| Category | Examples |
|---|---|
| Chronic infections | Tuberculosis, osteomyelitis, subacute bacterial endocarditis, HIV/AIDS, chronic fungal infection |
| Autoimmune/rheumatological | Rheumatoid arthritis (most common non-infective cause), SLE, IBD, vasculitis, sarcoidosis |
| Malignancy | Carcinomas, lymphomas, multiple myeloma (also direct BM involvement) |
| Chronic organ failure | Chronic kidney disease, heart failure, chronic liver disease, COPD |
| Trauma / Critical illness | Acute severe illness, post-surgical states, ICU patients |
Pathogenesis
ACD results from four interrelated mechanisms, all driven by inflammatory cytokines (IL-6, IL-1β, TNF-α, IFN-γ):
1. Hepcidin - The Master Regulator (Central Mechanism)
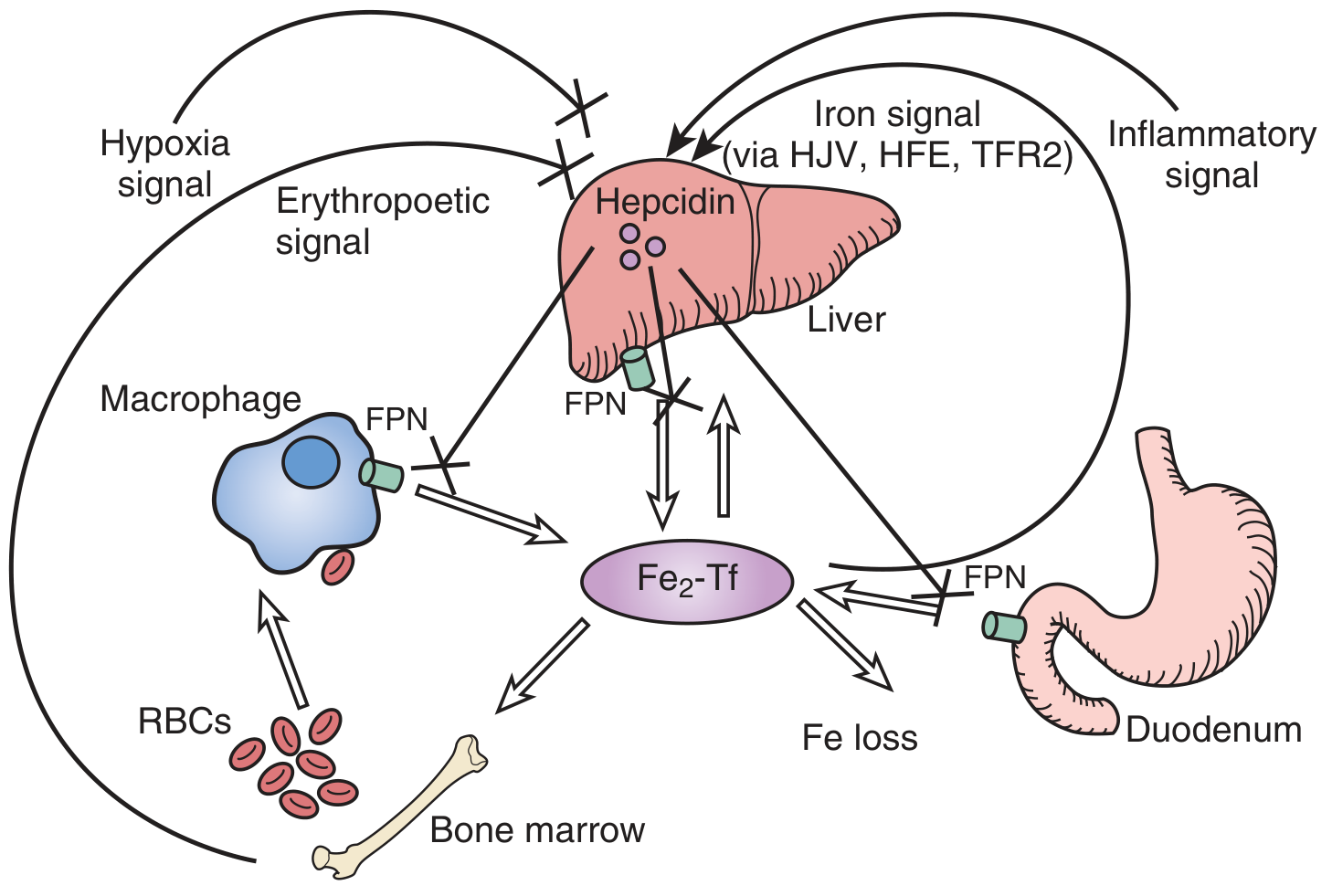
Fig. 55.6 (Brenner & Rector's The Kidney) - Hepcidin controls iron homeostasis via ferroportin degradation
Hepcidin is a 25-amino acid antimicrobial peptide produced and secreted by hepatocytes. It is the key hormone regulating systemic iron homeostasis:
Normal physiology:
- Iron overload → stimulates hepcidin (via HJV/hemojuvelin, HFE, TFR2 signalling → BMP/SMAD pathway → hepcidin gene transcription)
- Iron deficiency + hypoxia + erythropoietic drive → suppresses hepcidin → allows iron mobilisation
In ACD:
- Inflammatory cytokines, especially IL-6 → directly stimulate hepatic hepcidin synthesis (via JAK-STAT3 pathway)
- High hepcidin binds to and causes internalisation and degradation of ferroportin (FPN) - the only known mammalian iron exporter
- Ferroportin is expressed on:
- Duodenal enterocytes (iron absorption)
- RE macrophages (iron recycling from senescent RBCs)
- Hepatocytes (iron storage/release)
- Loss of ferroportin at all three sites results in:
- Blocked intestinal iron absorption
- Iron trapped in RE macrophages (functional iron deficiency)
- Reduced iron release from hepatocytes
- Result: serum iron falls despite replete (or increased) total body stores - "functional iron deficiency"
2. Reduced Erythropoietin (EPO) Production and Responsiveness
- Inflammatory cytokines (IL-1β, TNF-α, IFN-γ) directly suppress EPO gene transcription in peritubular renal cells
- Serum EPO is lower than expected for the degree of anaemia
- Additionally, EPO responsiveness of erythroid progenitors is blunted
- IFN-γ and TNF-α inhibit BFU-E and CFU-E colony formation in vitro
- Ceramide (a product of IFN-γ-induced sphingomyelin hydrolysis) acts as an intracellular mediator of erythroid progenitor inhibition
3. Direct Suppression of Erythropoiesis
- IFN-γ, TNF-α, IL-1β, and IL-13 directly inhibit erythroid progenitor proliferation and differentiation in bone marrow
- Reactive oxygen species generated at inflammatory sites damage erythroid precursors
- In malignancy - direct bone marrow infiltration by tumour cells
4. Shortened Red Cell Survival
- Increased macrophage activation (from inflammation) → accelerated erythrophagocytosis
- Mild extravascular haemolysis - RBC lifespan reduced from 120 days to ~60-90 days
- Cytokines alter RBC membrane deformability
- Iron-restricted erythropoiesis produces hypochromic, poorly deformable red cells
Integrated Pathogenesis Diagram
CHRONIC INFECTION / INFLAMMATION / MALIGNANCY
↓
┌─────────────────────────────────┐
│ Monocytes / Macrophages / T-cells│
│ Release: IL-6, IL-1β, TNF-α, │
│ IFN-γ │
└─────────────────────────────────┘
↓ ↓ ↓ ↓
IL-6 → Liver TNF-α/IFN-γ IFN-γ/TNF-α Macrophage
↑ Hepcidin ↓ EPO gene ↓ BFU-E/ activation →
↓ Ferroportin transcription CFU-E ↑ Erythro-
→ Fe trapped ↓ EPO levels → ↓ BM phagocytosis
in RE cells + ↓ response erythropoiesis → ↓ RBC survival
ALL → ANAEMIA
Morphological Features
Peripheral Blood
- Normocytic, normochromic anaemia in most cases (MCV 80-90 fL)
- Microcytic, hypochromic in 20-50% of cases (when hepcidin-mediated iron restriction is severe; MCV rarely <70 fL)
- Slight anisocytosis and poikilocytosis
- Reticulocyte count: not elevated (inappropriately low for degree of anaemia)
- Normal WBC and platelets (unless affected by underlying disease)
Bone Marrow
- Normocellular or mildly hypercellular/hypocellular
- Erythroid precursors: delayed Hb synthesis - frayed, hypochromic cytoplasm of late normoblasts
- Decreased sideroblasts (less iron available for developing RBCs)
- Macrophage iron stores normal or INCREASED - iron seen in macrophages on Perls' Prussian blue stain
This is a key distinguishing feature from IDA where macrophage stores are absent
Laboratory Findings
| Parameter | ACD | IDA | Combined ACD+IDA |
|---|---|---|---|
| Haemoglobin | 8-10 g/dL | Variable | Variable |
| MCV | Normal (normocytic) | Low (microcytic) | Low |
| Serum Iron | Low | Low | Low |
| TIBC (Transferrin) | Low or Normal | High | Normal or Low |
| Transferrin Saturation (TSAT) | Low (<16%) | Low | Low |
| Serum Ferritin | Normal or High (acute phase reactant) | Low (<12 μg/L) | Low-normal |
| Soluble Transferrin Receptor (sTfR) | Normal | Elevated | Elevated |
| sTfR / log Ferritin index | Low | High | High |
| Hepcidin | Elevated | Low | Variable |
| Serum EPO | Low for degree of anaemia | Appropriately elevated | Low |
| BM iron stores | Normal/Increased | Absent | Absent |
| Reticulocyte count | Inappropriately low | Inappropriately low | Low |
| ESR / CRP | Elevated | Normal | Elevated |
| Erythrocyte protoporphyrin | Elevated | Elevated | Elevated |
Key diagnostic clue: Low iron + Low TIBC + Normal/high ferritin + Normal/high BM stores = ACD Low iron + High TIBC + Low ferritin + Absent BM stores = IDA
Distinguishing ACD from Iron Deficiency Anaemia
The most challenging clinical problem is distinguishing ACD from true iron deficiency (or their coexistence):
| Test | ACD | IDA |
|---|---|---|
| Ferritin | >100 μg/L (usually) | <12 μg/L |
| sTfR | Normal | Elevated |
| sTfR/log Ferritin (Thomas index) | <1 | >2 |
| BM Perls' stain | Iron present in macrophages, absent in sideroblasts | No stainable iron |
| Response to IV iron | No improvement in Hb (unless co-deficient) | Hb rises |
| Hepcidin | High | Low |
Ferritin >100 μg/L essentially excludes iron deficiency (Goldman-Cecil); values of 12-100 μg/L are indeterminate and require sTfR or BM biopsy.
Clinical Features
- Anaemia is mild to moderate - Hb typically 8-10 g/dL; rarely <7 g/dL
- Symptoms: fatigue, pallor, exertional dyspnoea - often overshadowed by the underlying disease
- No specific signs (no koilonychia, no angular stomatitis - these suggest IDA)
- Symptoms of the underlying disease predominate
- Anaemia worsens with disease flares and improves with disease control
Treatment
Primary Approach: Treat the Underlying Disease
- The most effective therapy is control of the inflammatory disorder
- Treating infection, malignancy, or immunosuppressing the rheumatological disease normalises hepcidin and restores erythropoiesis
Specific Interventions
| Treatment | Indication |
|---|---|
| Erythropoiesis-stimulating agents (ESAs) - epoetin alfa, darbepoetin | ACD in CKD (standard of care); cancer patients on chemotherapy (with caution - thrombosis risk); NOT for non-chemotherapy cancer |
| Iron supplementation (oral or IV) | Only if concurrent true iron deficiency is present, OR in combination with ESA therapy (to prevent functional iron deficiency from increased erythropoietic demand) |
| Red cell transfusion | For symptomatic severe anaemia (Hb <7-8 g/dL) or haemodynamic compromise |
| Hepcidin antagonists (investigational/emerging) | Anti-hepcidin antibodies, anti-IL-6, anti-BMP/SMAD pathway agents - promising targets; roxadustat (PHD inhibitor) reduces hepcidin and increases EPO |
| Tocilizumab (anti-IL-6R) | Reduces hepcidin in RA patients; anaemia improves as a secondary benefit |
Summary - Key Concepts
| Concept | Detail |
|---|---|
| Nomenclature | ACD = Anaemia of Inflammation (preferred) |
| Central mediator | Hepcidin (25-aa liver peptide) |
| Hepcidin induction | IL-6 → JAK-STAT3 → hepcidin gene |
| Hepcidin effect | Degrades ferroportin → blocks Fe export from enterocytes, macrophages, hepatocytes |
| Net result | Functional iron deficiency - Fe trapped in RE system |
| BM stores | Normal/increased (differentiates from IDA) |
| TIBC | Low or normal (not elevated - differentiates from IDA) |
| EPO | Inadequately low for degree of anaemia |
| Treatment | Treat underlying cause; ESAs in CKD; hepcidin antagonists emerging |
Examiner's Scoring Guide
For 10 marks:
- Definition, nomenclature, epidemiology (second most common worldwide) - 1 mark
- Causes/associated conditions (infection, autoimmune, malignancy, organ failure) - 1 mark
- Pathogenesis - hepcidin mechanism (central), EPO suppression, direct erythroid suppression, shortened RBC survival - 3 marks
- Morphological features - PBS and BM - 1 mark
- Lab findings table (iron, TIBC, ferritin, sTfR, hepcidin) with comparison to IDA - 2.5 marks
- Treatment - 1.5 marks
Sources: Henry's Clinical Diagnosis and Management by Laboratory Methods; Brenner & Rector's The Kidney 11e; Goldman-Cecil Medicine International Edition; Tietz Textbook of Laboratory Medicine 7e
Classify myelodysplastic syndrome and mention diagnostic criteria in brief.
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.