Heart attack
Reading File
Asking for Preferences
Heart attack
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND management
Searching the Web
heart attack myocardial infarction overview 2025
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a thorough, textbook-based overview of heart attacks (myocardial infarction).
Heart Attack (Myocardial Infarction)
Definition
A myocardial infarction (MI) is the death of cardiac muscle due to prolonged ischemia - when blood flow to a portion of the myocardium is reduced or completely blocked for long enough to cause irreversible cell death. Roughly 800,000 people in the United States experience an MI each year (nearly one every 40 seconds), causing about 400,000 deaths annually. The underlying cause in the vast majority of cases is atherosclerotic coronary artery disease (CAD).
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 511
Causes & Pathogenesis
The Classic Sequence (90% of cases)
- An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents to circulating blood.
- Platelets adhere, aggregate, and activate, releasing thromboxane A2, ADP, and serotonin - causing further platelet aggregation and vasospasm.
- Coagulation is activated via tissue factor, adding to the growing thrombus.
- Within minutes, the thrombus can completely occlude the coronary artery lumen.
Angiography performed within 4 hours of MI onset demonstrates coronary thrombosis in almost 90% of cases. Notably, these occlusions most commonly occur at sites that did NOT previously have a critical (>70%) fixed stenosis - meaning a "mild" plaque can still cause a fatal MI if it ruptures suddenly.
Less Common Causes (~10% of cases)
- Vasospasm (with or without atherosclerosis) - triggered by cocaine, ephedrine, or platelet aggregation
- Embolism - from left atrial thrombus (in atrial fibrillation), infective endocarditis vegetations, or prosthetic cardiac material
- Small vessel disease - vasculitis, amyloid deposition
- Hematologic disorders - sickle cell disease, hypercoagulable states
The Progression of Necrosis
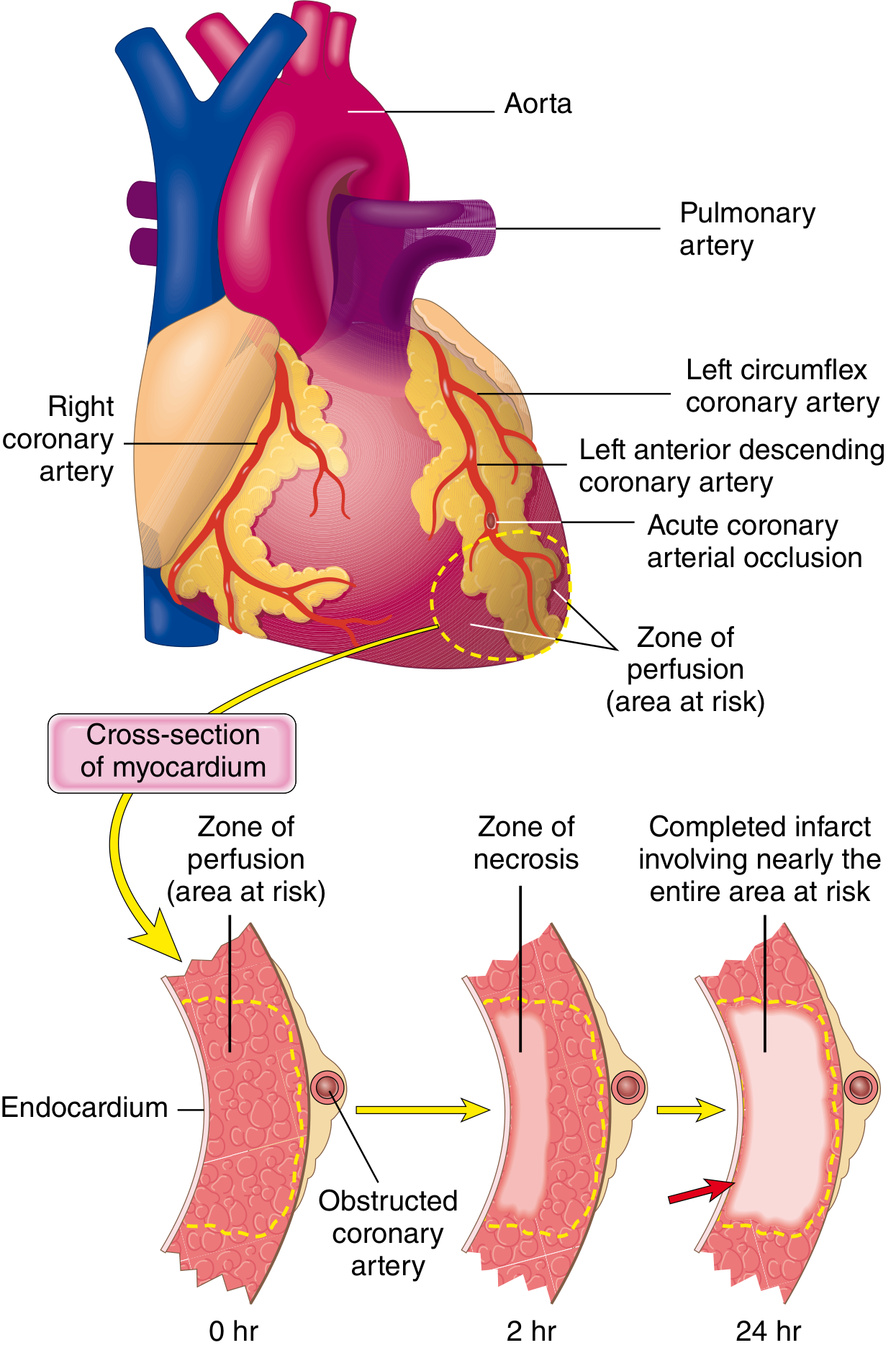
The timeline of cellular events is well defined:
| Event | Time After Occlusion |
|---|---|
| Onset of ATP depletion | Seconds |
| Loss of contractility | < 2 minutes |
| ATP reduced to 50% of normal | 10 minutes |
| Irreversible cell injury | 20-40 minutes |
| ATP reduced to 10% of normal | 40 minutes |
| Microvascular injury | > 1 hour |
Ischemic injury starts in the subendocardial zone (most vulnerable - last to receive blood, compressed by high intramural pressure during systole) and a wavefront of necrosis spreads outward toward the epicardium over hours. A full-thickness (transmural) infarct usually achieves its final size within 6-12 hours.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 512
- Guyton and Hall Textbook of Medical Physiology
Risk Factors
- Non-modifiable: Age, male sex (females gain similar risk post-menopause due to declining estrogen, rising cholesterol, blood pressure, and inflammatory markers), family history
- Modifiable: Hypertension, hypercholesterolemia, smoking, diabetes, obesity, physical inactivity
Symptoms
Classic presentation:
- Crushing, pressure-like chest pain - often radiating to the left arm, jaw, or shoulder
- Shortness of breath
- Diaphoresis (sweating), nausea, vomiting
- Sense of impending doom
Atypical/silent presentations occur in ~20-30% of patients - more common in diabetics and women - and may manifest only as fatigue, jaw pain, or epigastric discomfort.
Diagnosis
ECG Changes
Three major mechanisms cause ECG changes in acute MI:
| Defect in Infarcted Cells | Current Flow | ECG Change (leads over infarct) |
|---|---|---|
| Rapid repolarization | Out of infarct | ST segment elevation |
| Decreased resting membrane potential | Into infarct | TQ depression (appears as ST elevation) |
| Delayed depolarization | Out of infarct | ST segment elevation |
- STEMI (ST-Elevation MI): Full-thickness occlusion - a medical emergency requiring immediate reperfusion
- NSTEMI (Non-ST-Elevation MI): Partial occlusion or subendocardial infarction
- Q waves develop over days-weeks as dead tissue becomes electrically silent
Biomarkers
- Cardiac troponins (cTnI, cTnT): Most sensitive and specific - rise within 2-4 hours, peak at ~24 hours, remain elevated for up to 10-14 days
- CK-MB: Rises within 4-6 hours, returns to normal in ~48-72 hours (useful for detecting reinfarction)
Types of MI
- Type 1 MI: Spontaneous plaque rupture with thrombosis
- Type 2 MI: Supply-demand mismatch without plaque rupture (e.g., tachyarrhythmia, anemia, severe hypertension)
Treatment
Acute Management
- Aspirin immediately (antiplatelet)
- P2Y12 inhibitor (clopidogrel, ticagrelor, or prasugrel) - dual antiplatelet therapy
- Anticoagulation (heparin, bivalirudin)
- Nitroglycerin (sublingual then IV) - for pain and vasodilation
- Oxygen if SpO2 < 90%
- Morphine for refractory pain
Reperfusion (the core treatment)
- Primary PCI (percutaneous coronary intervention): The preferred strategy for STEMI - goal is door-to-balloon time < 90 minutes. A catheter is used to open the blocked artery with a balloon and stent.
- Fibrinolytic therapy (thrombolytics): Used when PCI is not available within 120 minutes of first medical contact. Examples: alteplase, tenecteplase.
- CABG (coronary artery bypass grafting): Reserved for multivessel disease or anatomical patterns not suitable for PCI.
Long-Term Medications
- Beta-blockers - reduce heart rate, myocardial oxygen demand, and mortality
- ACE inhibitors/ARBs - reduce remodeling, especially if ejection fraction is reduced
- Statins - intensive statin therapy (e.g., atorvastatin 80 mg) to reduce LDL and plaque progression
- Dual antiplatelet therapy - continued for at least 12 months post-stent
Complications
| Complication | Timing | Notes |
|---|---|---|
| Arrhythmias (VF, VT, heart block) | Immediately | Leading early cause of death |
| Cardiogenic shock | Hours-days | Pump failure; high mortality |
| Pulmonary edema | Hours-days | Left ventricular failure causes back-pressure |
| Pericarditis | Days 2-4 (early) | Fibrinous; Dressler syndrome weeks later |
| Papillary muscle rupture | Days 3-5 | Causes acute mitral regurgitation |
| Ventricular free wall rupture | Days 3-5 | Catastrophic; causes cardiac tamponade |
| Ventricular septal defect | Days 3-5 | New harsh murmur; hemodynamic compromise |
| Left ventricular aneurysm | Weeks-months | Risk of mural thrombus and systemic emboli |
| Reinfarction | Any time | Higher risk with non-Q-wave (subendocardial) infarcts |
Regarding causes of death, the four major mechanisms are: (1) decreased cardiac output / cardiogenic shock (the infarcted zone may "bulge out" during systole - called systolic stretch - losing its pumping contribution); (2) pulmonary edema; (3) ventricular fibrillation; and (4) cardiac rupture.
- Guyton and Hall Textbook of Medical Physiology
Prevention
- Smoking cessation
- Blood pressure control (target < 130/80 mmHg)
- Lipid lowering (statin therapy for high-risk individuals)
- Diabetes management
- Regular physical activity
- Healthy diet (Mediterranean-style)
- Aspirin in selected high-risk patients (shared decision-making)
Current Guidelines
The 2025 ACC/AHA/ACEP/NAEMSP/SCAI Guideline for the Management of Patients With Acute Coronary Syndromes (published April 2025 in Circulation, PMID 40014670) is the most current comprehensive guideline covering the full spectrum from pre-hospital care through long-term secondary prevention.
A 2025 systematic review on beta-blockers after MI (PMID 39298680) found that for patients without reduced ejection fraction or heart failure, the mortality benefit of long-term beta-blockers after MI is less clear than previously thought - an evolving area.
Summary: A heart attack is a time-critical emergency. The key principle is "time is muscle" - every minute of delay means more cardiomyocytes dying. Rapid recognition, early activation of emergency services, and prompt reperfusion (ideally within 90 minutes for STEMI) are the most important determinants of outcome.
This is a shared conversation. Sign in to Orris to start your own chat.