Drugs used in ophthalmology from basic to advanced level; include pharmacodynamics, phar
Drugs used in ophthalmology from basic to advanced level; include pharmacodynamics, pharmacokinetics, uses, side effects, preservatives used, how it is prepared, any clinical trails and recent advances everything. Possible every mechanisms available
"ophthalmic drugs" AND "pharmacology"
ophthalmic pharmacology drugs ophthalmology complete pharmacodynamics pharmacokinetics 2024 2025
"anti-VEGF" AND "ophthalmic" AND "intravitreal"
"glaucoma" AND "pharmacotherapy" AND "prostaglandin"
"ophthalmic drug delivery" AND "recent advances" AND "nanoparticles"
"faricimab" AND "retinal" AND "clinical trial"
Drugs Used in Ophthalmology - Complete Pharmacology Reference
Table of Contents
- Ocular Pharmacokinetics - Barriers & Drug Delivery
- Miotics (Cholinergic Agonists)
- Mydriatics & Cycloplegics (Muscarinic Antagonists)
- Sympathomimetics
- Beta-Adrenergic Blockers (Antiglaucoma)
- Alpha-2 Agonists (Antiglaucoma)
- Prostaglandin Analogs
- Carbonic Anhydrase Inhibitors
- Rho Kinase (ROCK) Inhibitors
- Osmotic Agents
- Topical Anesthetics
- Anti-infective Agents (Antibacterials, Antivirals, Antifungals, Antiparasitics)
- Anti-inflammatory Agents (Steroids, NSAIDs)
- Antiallergic / Antihistamine Agents
- Anti-VEGF Biologics
- Immunosuppressants for Dry Eye
- Surgical Adjuncts
- Dyes & Diagnostic Agents
- Ocular Toxicity of Systemic Drugs
- Preservatives in Ophthalmic Formulations
- Recent Advances & Novel Drug Delivery
1. OCULAR PHARMACOKINETICS - BARRIERS & DRUG DELIVERY
Routes of Administration
| Route | Examples | Bioavailability Notes |
|---|---|---|
| Topical drops | Most antiglaucoma, antibiotics | <5% systemic absorption; nasolacrimal drainage causes systemic side effects |
| Subconjunctival | Corticosteroids, antibiotics | Bypasses corneal barrier; good for anterior segment |
| Intravitreal | Anti-VEGF, antibiotics | Direct vitreous delivery; best posterior segment access |
| Periocular (sub-Tenon's) | Triamcinolone | Good for posterior uveitis |
| Systemic (oral/IV) | Acetazolamide, mannitol | Useful for posterior segment and acute glaucoma |
Ocular Barriers to Drug Penetration
- Corneal epithelium: Tight junctions limit hydrophilic drug penetration; lipophilic drugs penetrate better
- Blood-aqueous barrier: Tight junctions in ciliary epithelium and iris vessels
- Blood-retinal barrier: Inner (retinal endothelium) and outer (RPE) layers
- Nasolacrimal drainage: Topical drugs drain rapidly, reducing bioavailability and causing systemic absorption via nasal mucosa
- Tear film turnover: ~16% per minute - reduces contact time; only about 5-10 µL of a standard 30-50 µL drop is retained
Pharmacokinetic Factors
- Vehicle/formulation: Gel-forming drops (Timoptic-XE), viscosity enhancers (HPMC), ointments, and sustained-release inserts all increase contact time
- Protein binding: Affects distribution in aqueous humor
- Melanin binding: Drugs like atropine, chloroquine bind to melanin in uveal tissue, creating a depot effect and prolonging action (also source of pigment-related toxicity)
2. MIOTICS (PARASYMPATHOMIMETICS - CHOLINERGIC AGONISTS)
Direct-Acting Miotics
Pilocarpine
- Class: Muscarinic receptor agonist (M3)
- Pharmacodynamics:
- Contracts iris sphincter muscle → miosis (M3 receptor in iris sphincter)
- Contracts ciliary muscle → accommodation (spasm)
- In glaucoma: ciliary muscle contraction opens trabecular meshwork by pulling on scleral spur → increases aqueous outflow (conventional/trabecular pathway)
- Lowers IOP by 20-30%
- Pharmacokinetics:
- Topical 1-4% solution or 4% gel
- Onset: 10-30 min; Duration: 4-8 hours (gel: once-daily dosing)
- Ocusert (pilocarpine-impregnated membrane) provides sustained 7-day release
- Systemic absorption: minimal at therapeutic doses
- Uses: Open-angle glaucoma (now 4th-line), acute angle-closure glaucoma (emergency), reversal of mydriasis after examination, esotropia (occasionally)
- Side effects: Induced myopia (ciliary spasm), brow ache/headache, decreased night vision (miosis), retinal detachment risk (vitreous traction), systemic - nausea, salivation, diaphoresis (rare)
Carbachol
- Class: Cholinomimetic (muscarinic + nicotinic); resistant to cholinesterase
- Pharmacodynamics: Similar to pilocarpine; also stimulates nicotinic receptors at ganglia
- Uses: Intraocular miosis during cataract/glaucoma surgery (0.01% intraocular); topical glaucoma (rarely now)
- Formulation: 0.01% intraocular solution (preservative-free, Miostat)
Indirect-Acting Miotics (Cholinesterase Inhibitors)
Echothiophate Iodide (Phospholine Iodide)
- Class: Irreversible organophosphate AChE inhibitor
- Pharmacodynamics: Irreversibly phosphorylates active site serine of AChE → massive accumulation of ACh at all muscarinic and nicotinic synapses
- Uses: Accommodative esotropia in children; occasionally refractory glaucoma
- Side effects: Iris cysts (especially in children - prevented by phenylephrine co-administration), cataracts (with prolonged use), systemic cholinergic toxicity if absorbed, contraindicated before succinylcholine (risk of prolonged apnea - AChE inhibition extends duration of succinylcholine)
- ANESTHETIC ALERT: Must be discontinued 4-6 weeks before general anesthesia with succinylcholine
3. MYDRIATICS & CYCLOPLEGICS (MUSCARINIC ANTAGONISTS)
| Drug | Onset | Duration | Cycloplegia | Primary Use |
|---|---|---|---|---|
| Atropine | 30-40 min | 7-14 days | Complete | Uveitis, penalization therapy |
| Scopolamine (hyoscine) | 20-30 min | 5-7 days | Nearly complete | Uveitis |
| Homatropine | 40-60 min | 1-3 days | Good | Uveitis, refraction |
| Cyclopentolate | 30-60 min | 12-24 hrs | Excellent | Refraction (gold standard in children) |
| Tropicamide | 20-30 min | 4-6 hrs | Partial | Fundus examination |
| Phenylephrine (alpha agonist) | 15-20 min | 3-5 hrs | None | Mydriasis only, breaks posterior synechiae |
Mechanism Summary
- Iris sphincter: M3 receptor blockade → pupil dilation (mydriasis)
- Ciliary muscle: M3 receptor blockade → relaxation → loss of accommodation (cycloplegia)
- Aqueous humor: Slight reduction in production (incidental)
Pharmacokinetics
- Topical absorption through cornea; systemic absorption via nasolacrimal duct
- Duration varies by lipophilicity and receptor binding affinity
- Melanin binding prolongs effect in darkly pigmented eyes
Side Effects
- Ocular: photophobia, blurred near vision, raised IOP in narrow-angle glaucoma (angle closure precipitation - EMERGENCY)
- Systemic (especially atropine in children): tachycardia, flushing, dry mouth, fever, confusion, urinary retention, "hot as a hare, dry as a bone, red as a beet, blind as a bat, mad as a hatter"
- Tropicamide lowest systemic risk; Atropine highest due to long duration
Uses
- Diagnostic: Fundus examination, refraction
- Therapeutic: Uveitis (prevent posterior synechiae, relieve ciliary spasm), amblyopia therapy (atropine penalization)
- Surgical: Pre-/intraoperative mydriasis
4. SYMPATHOMIMETICS
Phenylephrine (Alpha-1 Agonist)
- Pharmacodynamics: Selective α1 agonist → contracts iris dilator muscle (mydriasis without cycloplegia)
- Uses: Diagnostic mydriasis, combined with cycloplegics to break posterior synechiae in uveitis, decongestant in OTC eye drops
- Concentrations: 2.5% (usual), 10% (reserved for adults; risk of hypertensive crisis)
- Side Effects: Systemic absorption with 10% = hypertensive crisis, reflex bradycardia, arrhythmia; avoid in cardiac disease
- Pharmacokinetics: Rapid onset (15-20 min), duration 3-5 hours
Epinephrine / Dipivefrin
- Dipivefrin: Prodrug of epinephrine (dipivalyl ester); better corneal penetration; converted to epinephrine by corneal esterases
- Mechanism: α + β adrenergic effects → decreases aqueous production (β2) + increases outflow (α1, trabecular and uveoscleral)
- Status: Largely replaced by newer agents due to side effects (cystoid macular edema in aphakic eyes, local reactions)
5. BETA-ADRENERGIC BLOCKERS (ANTIGLAUCOMA)
Mechanism
- No effect on aqueous outflow
- Lower IOP by 20-30%
Non-Selective (β1 + β2 Blockers)
| Drug | Concentration | Dosing |
|---|---|---|
| Timolol maleate | 0.25%, 0.5% | BD or Timoptic-XE (gel, OD) |
| Levobunolol | 0.25%, 0.5% | OD or BD |
| Metipranolol | 0.3% | BD |
| Carteolol | 1%, 2% | BD; intrinsic sympathomimetic activity |
Selective (β1 Blocker)
| Drug | Concentration | Notes |
|---|---|---|
| Betaxolol | 0.25%, 0.5% | β1 selective; safer in asthma; slightly less IOP-lowering but may have neuroprotective effect (Ca2+ channel blocking) |
Pharmacokinetics
- Well absorbed through cornea; significant nasolacrimal drainage causes systemic absorption
- Timolol half-life: ~4 hours (plasma); IOP effect 12-24 hrs
Adverse Effects - CRITICAL
- Pulmonary: Bronchospasm (potentially fatal in asthma/COPD) - even β1-selective drugs have risk
- Cardiovascular: Bradycardia, AV block, hypotension, heart failure exacerbation - contraindicated in 2nd/3rd degree AV block, severe bradycardia, overt cardiac failure
- CNS: Depression, fatigue, sexual dysfunction, insomnia
- Metabolic: Masks hypoglycemia symptoms in diabetics; prolongs hypoglycemia; alters lipid profile
- Ocular: Dry eye, punctate keratitis
Contraindications
- Asthma, COPD (all beta-blockers)
- Sinus bradycardia, AV block, cardiac failure
- Depression (relative)
- Infants and neonates (brimonidine preferred)
6. ALPHA-2 ADRENERGIC AGONISTS
Brimonidine (0.1%, 0.15%, 0.2%)
- Mechanism: Selective α2 agonist →
- Reduces aqueous humor production (via inhibition of cAMP in ciliary epithelium)
- Increases uveoscleral outflow
- Possible neuroprotective effect (independent of IOP)
- IOP reduction: ~20-25%
- Pharmacokinetics: Crosses blood-brain barrier; peak aqueous humor concentration at 1-2 hrs; systemic half-life ~3 hrs
- Side effects: Ocular allergy (10-20%), conjunctival follicles, drowsiness, dry mouth, headache, fatigue; potentially CNS depression and apnea in neonates/infants - CONTRAINDICATED in children <2 years and premature infants (risk of fatal CNS depression/apnea)
- Advantage over apraclonidine: Less tachyphylaxis, fewer allergic reactions, less systemic alpha-1 effects (no significant pupil dilation)
- Drug interactions: MAO inhibitors (avoid), TCAs (reduce efficacy), CNS depressants (additive)
Apraclonidine (0.5%, 1%)
- Less selective α2, also has α1 activity
- Mainly used short-term perioperatively (prevents IOP spike after laser procedures)
- Higher allergy rate (30-50% long-term) - not suitable for chronic therapy
- Reduces aqueous production primarily
7. PROSTAGLANDIN ANALOGS (PROSTAMIDES)
Mechanism
- Act at FP receptors (prostaglandin F receptors) on ciliary muscle
- Increase uveoscleral outflow (unconventional pathway) - relax ciliary muscle extracellular matrix, upregulate matrix metalloproteinases (MMPs) → remodel tissue → increased flow through ciliary body face and suprachoroidal space
- Some also increase trabecular outflow slightly
- IOP reduction: 25-35% (most powerful class)
- Once-daily evening dosing (maximum effect during night/morning when IOP tends to peak)
Agents
| Drug | Formulation | FP Receptor Activity | Notes |
|---|---|---|---|
| Latanoprost | 0.005% drops | Prodrug → free acid metabolite | First-in-class (1996) |
| Bimatoprost | 0.01%, 0.03% | Prostamide + FP receptor | Also FDA-approved for eyelash hypotrichosis (Latisse 0.03%) |
| Travoprost | 0.004% | FP receptor prodrug | Similar to latanoprost |
| Tafluprost | 0.0015% | FP prodrug | Available preservative-free |
| Unoprostone | 0.15% | Weaker FP + K+ channel | Less IOP lowering |
Pharmacokinetics
- Prodrugs (except bimatoprost which is a prostamide): hydrolyzed by corneal esterases to active free acid
- Onset: 3-4 hours; peak at 8-12 hours; duration 24 hours
- Minimal systemic exposure from once-daily topical use
- Active acid enters systemic circulation, metabolized hepatically
Side Effects (Unique to Class)
- Iris heterochromia: Irreversible increase in brown iris pigmentation (increased melanin synthesis in iris melanocytes) - particularly in green-hazel or blue/grey-brown irides; occurs in ~10% over years
- Periorbital changes (prostaglandin-associated periorbitopathy - PAP): Upper eyelid deepening/sulcus, lower lid fat atrophy, enophthalmos, ptosis - caused by periorbital fat atrophy
- Eyelash changes: Increased length, thickness, number, and pigmentation (trichomegaly)
- Conjunctival hyperemia (bimatoprost > others)
- Reactivation of herpes simplex keratitis
- Macular edema in aphakic/pseudophakic patients - use with caution
- Systemic: flu-like symptoms (rare), joint/back pain
8. CARBONIC ANHYDRASE INHIBITORS (CAIs)
Mechanism
- Carbonic anhydrase (CA) in non-pigmented ciliary epithelium catalyzes: HCO₃⁻ + H⁺ ↔ H₂O + CO₂ (via CA-II and CA-XII isoforms)
- Inhibition of CA reduces bicarbonate secretion → reduces Na+/H₂O transport into posterior chamber → reduces aqueous production by 30-50%
- Does NOT affect outflow
Topical CAIs
| Drug | Concentration | Dosing | Notes |
|---|---|---|---|
| Dorzolamide | 2% | BD-TDS | First topical CAI (1995) |
| Brinzolamide | 1% | BD-TDS | Less stinging; suspension |
- Pharmacokinetics: Penetrate corneal epithelium; distribute to red blood cells (bind to CA in RBCs) → long half-life in blood; renal excretion
- Side effects: Ocular stinging/burning (dorzolamide > brinzolamide), bitter taste, superficial punctate keratitis, allergic reactions; systemic effects rare but possible (sulfonamide allergy - cross-reactivity)
- Contraindication: Sulfonamide allergy; severe renal impairment
Oral/Systemic CAIs
| Drug | Dose | Notes |
|---|---|---|
| Acetazolamide | 125-250 mg BD-QID; 500 mg SR BD | Most commonly used; for acute angle closure, preoperative IOP reduction |
| Methazolamide | 25-50 mg BD-TDS | Better tolerated than acetazolamide |
| Dichlorphenamide | Rarely used |
- Side effects (oral): Paresthesias (hands/feet), malaise, fatigue, anorexia, depression, nephrolithiasis (calcium oxalate stones), metabolic acidosis, aplastic anemia (rare but serious), hypokalemia; Stevens-Johnson syndrome (rare)
- Contraindications: Sulfonamide hypersensitivity, adrenal insufficiency, hyponatremia/hypokalemia, hepatic cirrhosis, sickle cell disease
9. RHO KINASE (ROCK) INHIBITORS
Netarsudil (Rhopressa, 0.02%)
- Newest class of antiglaucoma drugs (FDA approved 2017)
- Mechanism (triple action):
- Rho kinase inhibition: Relaxes trabecular meshwork contractile cells → increases conventional (trabecular) outflow
- Norepinephrine transporter (NET) inhibition: Reduces sympathetic tone in ciliary body → reduces aqueous production
- Reduces episcleral venous pressure: Contributes to IOP lowering
- IOP reduction: ~20-25%; once-daily dosing
- Unique advantage: Works on trabecular meshwork (unlike prostaglandins which act on uveoscleral route) - can be additive with PGAs
- Pharmacokinetics: Undergoes intracorneal esterase hydrolysis to active metabolite AR-13503; rapid onset
- Side effects: Conjunctival hyperemia (most common, ~50%), subconjunctival hemorrhage, corneal verticillata (gold-brown corneal deposits - dose-dependent, reversible), blepharitis, increased tearing, pain
Roclatan (Netarsudil 0.02% + Latanoprost 0.005%)
- Fixed-dose combination; once daily
- Additive ~8-9 mmHg IOP reduction (MERCURY 1 & 2 trials)
10. OSMOTIC AGENTS
Mechanism
- Create osmotic gradient between plasma and vitreous/aqueous → draw water out of the eye → rapidly reduce IOP
- Used for acute IOP elevation, preoperative vitreous dehydration
| Drug | Route | Dose | Duration | Notes |
|---|---|---|---|---|
| Mannitol | IV | 1-2 g/kg over 30-60 min | 3-6 hrs | Drug of choice for acute angle closure (non-oral); contraindicated in cardiac/renal failure |
| Glycerin | Oral | 1-1.5 g/kg | 3-5 hrs | Metabolized to glucose; use with caution in diabetes |
| Isosorbide | Oral | 1.5 g/kg | — | Safer in diabetics than glycerin; not metabolized |
| Urea | IV | Rarely used now | — | Higher CNS penetration, more rebound |
- Side effects: Headache (volume shift), nausea/vomiting, electrolyte disturbances, pulmonary edema (heart failure), urinary retention (urologist alert); acute tubular necrosis with mannitol in renal failure
11. TOPICAL OCULAR ANESTHETICS
Mechanism
- Block voltage-gated Na+ channels → stabilize neuronal membrane → prevent action potential propagation in sensory corneal/conjunctival nerves
- No effect on pupil or IOP
| Drug | Concentration | Onset | Duration | Uses |
|---|---|---|---|---|
| Proparacaine (proxymetacaine) | 0.5% | 20-30 sec | 10-15 min | Tonometry, foreign body removal, minor procedures |
| Tetracaine | 0.5% | 30-60 sec | 15-20 min | More potent; diagnostic/procedural |
| Benoxinate | 0.4% | — | — | Combined with fluorescein |
| Lidocaine | 2-4% | Rapid | Variable | Peribulbar/retrobulbar blocks for surgery; intracameral use |
| Cocaine | 4-10% | — | 30-45 min | Also causes mydriasis (alpha agonist); limited to ENT/ophthalmology ENT procedures |
Important Notes
- Topical anesthetics are NOT to be dispensed for home use: Repeated use causes corneal epithelial toxicity, delayed healing, stromal infiltrates, corneal ulceration - suppress pain so patients ignore injury
- Used only in office/surgical setting
- Local infiltration/nerve blocks: Bupivacaine (long-acting), lidocaine used for retrobulbar/peribulbar anesthesia for intraocular surgery
12. ANTI-INFECTIVE OPHTHALMIC AGENTS
12a. Antibacterials
Fluoroquinolones (Preferred for bacterial keratitis and endophthalmitis prophylaxis)
- Mechanism: Inhibit bacterial DNA gyrase (topoisomerase II) and topoisomerase IV → prevent DNA replication, transcription, repair
- Spectrum: Broad gram-positive and gram-negative; some anaerobic activity (moxifloxacin)
| Drug | Generation | Concentration | Notes |
|---|---|---|---|
| Ciprofloxacin | 2nd | 0.3% drops, 0.3% ointment | Good P. aeruginosa; Staph coverage lower |
| Ofloxacin | 2nd | 0.3% | Broad spectrum |
| Levofloxacin | 3rd | 0.5% | Better gram-positive coverage |
| Moxifloxacin | 4th | 0.5% (Vigamox) | Best Streptococcus & MRSA coverage; no preservative needed (self-preserved) |
| Besifloxacin | 4th | 0.6% | Ophthalmic-only fluoroquinolone; no systemic use = reduced resistance pressure |
| Gatifloxacin | 4th | 0.3%, 0.5% | Broad spectrum |
Aminoglycosides
- Mechanism: Bind 30S ribosomal subunit → inhibit protein synthesis; bactericidal
- Tobramycin 0.3%: Excellent gram-negative coverage (Pseudomonas); combined with dexamethasone (Tobradex)
- Gentamicin 0.3%: Similar spectrum; more corneal toxicity
- Neomycin: Combined preparations; high allergy rate (10-15%)
Macrolides
- Azithromycin 1% (AzaSite): Inhibits 50S subunit; good for Chlamydia trachomatis (trachoma), blepharitis, bacterial conjunctivitis; advantages: once-daily dosing, prolonged retention in ocular tissues
Chloramphenicol
- Broad spectrum; bacteriostatic (binds 50S, inhibits peptidyl transferase)
- Risk of aplastic anemia (rare, dose-independent, idiosyncratic) - limits systemic use; topical ophthalmic use considered acceptable
- Available as eye drops and ointment; widely used in UK
Tetracyclines
- Tetracycline ointment: Trachoma (WHO recommended), Chlamydia, rosacea blepharitis
- Doxycycline (oral): Meibomian gland dysfunction, ocular rosacea - modulates MMP activity, reduces inflammatory cytokines
Polymyxin B Combinations
- Polymyxin B + trimethoprim (Polytrim): Broad coverage; first-line for bacterial conjunctivitis (children)
- Polymyxin B + neomycin + gramicidin/bacitracin (Neosporin): Classic broad-spectrum; common sensitizer
12b. Antiviral Agents
| Drug | Mechanism | Virus Target | Formulation | Use |
|---|---|---|---|---|
| Trifluridine (trifluorothymidine) | Thymidine analog; inhibits DNA polymerase | HSV-1, HSV-2 | 1% drops | Herpetic keratitis; limited by toxicity |
| Acyclovir | Guanosine analog; phosphorylated by viral thymidine kinase → inhibits viral DNA pol | HSV | 3% ointment; oral | Herpetic keratitis; oral for dendritic ulcers, iritis |
| Ganciclovir | Acyclovir prodrug-like; phosphorylated by UL97 kinase in CMV | CMV, HSV | 0.15% gel; IV; intravitreal implant | CMV retinitis (intravitreal implant Vitrasert), HSV keratitis |
| Valganciclovir | Oral prodrug of ganciclovir | CMV | Oral | CMV retinitis |
| Foscarnet | Pyrophosphate analog; directly inhibits viral DNA pol (no phosphorylation needed) | CMV, HSV (acyclovir-resistant) | Intravitreal injection | Resistant CMV/HSV retinitis |
| Cidofovir | dCMP analog; inhibits viral DNA pol | CMV | Intravitreal (20 μg); topical | CMV retinitis |
| Fomivirsen | Antisense oligonucleotide; blocks CMV mRNA | CMV | Intravitreal | CMV retinitis (orphan drug; withdrawn in some markets) |
Herpetic Eye Disease
- Dendritic corneal ulcer (HSV): Topical acyclovir ointment 5x/day or ganciclovir 0.15% gel 5x/day; debridement
- HSV stromal keratitis: Add topical steroids (with antiviral cover) to reduce immune-mediated stromal damage
- Herpes Zoster Ophthalmicus: Oral acyclovir/valacyclovir/famciclovir within 72h; prevents ocular complications
12c. Antifungal Agents
| Drug | Class | Mechanism | Formulation | Use |
|---|---|---|---|---|
| Natamycin | Polyene | Binds ergosterol → disrupts fungal membrane permeability | 5% suspension (only FDA-approved topical antifungal) | Filamentous fungal keratitis (Aspergillus, Fusarium) |
| Amphotericin B | Polyene | Binds ergosterol → membrane disruption | 0.1-0.5% topical (compounded); intravitreal 5-10 µg | Candida keratitis/endophthalmitis |
| Voriconazole | Triazole | Inhibits CYP51 (lanosterol 14α-demethylase) → inhibits ergosterol synthesis | Oral, IV; intravitreal 50 µg | Broad antifungal; good for resistant fungi |
| Fluconazole | Triazole | CYP51 inhibition | Oral, IV | Candida |
| Itraconazole | Triazole | CYP51 inhibition | Oral | Filamentous fungi |
- Source: Goodman & Gilman's, Table 74-6
12d. Antiparasitic Agents
- Acanthamoeba keratitis: Topical propamidine isethionate (Brolene) + polyhexamethylene biguanide (PHMB) or chlorhexidine (both compounded); adjunct: oral miltefosine (FDA-approved for leishmaniasis; off-label)
- Toxoplasmosis chorioretinitis: Pyrimethamine + sulfadiazine + folinic acid (leucovorin) ± clindamycin ± oral steroids; alternative: TMP-SMX ± clindamycin
- Onchocerciasis (river blindness): Ivermectin (oral) - kills microfilariae
13. ANTI-INFLAMMATORY AGENTS
13a. Corticosteroids
Mechanism
- Bind intracellular glucocorticoid receptors (GRs) → GR-drug complex translocates to nucleus → binds glucocorticoid response elements (GREs) → upregulates anti-inflammatory proteins (lipocortin/annexin-1, IL-10)
- Inhibits phospholipase A2 (via annexin-1) → reduces arachidonic acid release → reduces prostaglandins + leukotrienes
- Reduces vascular permeability, inhibits leukocyte migration, suppresses cytokine production
- Stabilizes lysosomal membranes
Potency Classification (Ophthalmic)
- Prednisolone acetate 1% (Pred Forte): Gold standard; suspension; must be shaken well
- Dexamethasone 0.1%: High potency; phosphate form better penetration
- Difluprednate 0.05% (Durezol): Difluorinated; potent; less frequent dosing; higher IOP risk
- Fluorometholone 0.1%, 0.25% (FML): Less IOP elevation and cataract risk; less penetration; good for conjunctival disease
- Medrysone 1%: Lowest penetration; limited to conjunctival/allergic use
- Loteprednol etabonate 0.2%, 0.5% (Lotemax, Alrex): "Retrometabolic" design; undergoes predictable inactivation after receptor binding; significantly less IOP elevation and cataract risk; now available as 0.5% gel and 1% suspension
- Rimexolone 1% (Vexol): For post-op inflammation, anterior uveitis
Uses
- Anterior uveitis/iritis, allergic conjunctivitis (severe), post-surgical inflammation, corneal graft rejection, vernal keratoconjunctivitis, episcleritis, scleritis, inflammatory conditions of posterior segment (periocular/intravitreal)
Routes in Posterior Segment
- Sub-Tenon's triamcinolone 40 mg: Posterior uveitis, CMO
- Intravitreal triamcinolone 4 mg: DME, BRVO-associated CMO; reactivates latent ocular infections
- Ozurdex (dexamethasone intravitreal implant 0.7 mg): Biodegradable PLGA implant; ~6 months duration; approved for DME, BRVO/CRVO, non-infectious posterior uveitis
- Iluvien (fluocinolone acetonide implant 0.19 mg): Non-biodegradable; ~36 months duration; approved for chronic DME
Adverse Effects - Ocular (Dose and Duration Dependent)
- Raised IOP (steroid glaucoma): Mechanism - steroids increase trabecular meshwork glycosaminoglycans → reduced outflow; affects ~30% of general population ("steroid responders"); can lead to irreversible glaucoma
- Posterior subcapsular cataracts (PSC): With prolonged topical or systemic use; mechanism relates to inhibition of lens epithelial cell differentiation
- Infection activation/worsening: Herpes simplex, fungal, bacterial; never use steroids alone on "red eye" without diagnosis
- Impaired wound healing
- Increased risk of perforation in corneal ulcers
13b. NSAIDs (Ophthalmic)
Mechanism
- Inhibit cyclooxygenase (COX-1 and COX-2) → reduce prostaglandin synthesis in ocular tissues → reduce inflammation and pain without IOP elevation
| Drug | Concentration | COX Selectivity | Uses |
|---|---|---|---|
| Diclofenac | 0.1% | COX-1 > COX-2 | Post-cataract CME prevention, corneal analgesia |
| Ketorolac | 0.4%, 0.5% | Non-selective | Post-op pain/inflammation, seasonal allergic conjunctivitis |
| Nepafenac | 0.1%, 0.3% | Prodrug → amfenac | Post-cataract CME; better penetration as prodrug |
| Bromfenac | 0.07%, 0.09%, 0.1% | COX-2 > COX-1 | Post-op inflammation; once-daily (0.07%); also studied in VEGF-driven maculopathies (meta-analysis 2024 PMID 39180057) |
| Flurbiprofen | 0.03% | Non-selective | Intraoperative miosis prevention; maintain surgical mydriasis |
| Suprofen | 1% | Non-selective | Intraoperative miosis prevention |
Adverse Effects
- Corneal toxicity/melting (prolonged use post-keratorefractive surgery - especially diclofenac)
- Superficial punctate keratitis, burning/stinging
- Delay epithelial healing
- Precipitation of angle closure (via prostaglandin inhibition)
14. ANTIALLERGIC / ANTIHISTAMINE AGENTS
H1 Antihistamines with Mast Cell Stabilizers (Dual Action - Preferred)
| Drug | Receptor Action | Concentration | Dosing |
|---|---|---|---|
| Olopatadine | H1 + mast cell stabilizer | 0.1% (BD), 0.2% (OD), 0.7% (OD) | Gold standard; most prescribed |
| Ketotifen | H1 + mast cell stabilizer | 0.025% | OTC available |
| Bepotastine | H1 + mast cell stabilizer | 1.5% | BD; also inhibits eosinophil migration |
| Alcaftadine | H1 + H2 + mast cell stabilizer | 0.25% | OD; blocks H1 AND H2; reduces eosinophil transmigration |
| Azelastine | H1 + mast cell stabilizer | 0.05% | BD |
| Epinastine | H1 + H2 + mast cell stabilizer | 0.05% | BD |
Pure Mast Cell Stabilizers
| Drug | Mechanism | Use |
|---|---|---|
| Cromolyn sodium | Stabilizes mast cell membranes (prevents degranulation); must be used regularly (4x/day) | Vernal/atopic conjunctivitis; effective only prophylactically |
| Nedocromil | Mast cell stabilizer + some antihistamine | Vernal keratoconjunctivitis |
| Lodoxamide | More potent mast cell stabilizer; inhibits eosinophil chemotaxis | Vernal keratoconjunctivitis |
| Pemirolast | Mast cell stabilizer | Allergic conjunctivitis |
Pure H1 Antihistamines (Topical, Older)
- Levocabastine 0.05% (highly selective H1; minimal mast cell effect)
- Emedastine 0.05%
Pharmacodynamics of Mast Cell Stabilizers
- Inhibit IgE-mediated calcium influx into mast cells → prevent degranulation → reduce release of histamine, prostaglandins, leukotrienes, cytokines
- Must be started BEFORE allergen exposure; not effective for immediate relief
15. ANTI-VEGF BIOLOGICS (INTRAVITREAL)
Vascular Endothelial Growth Factor (VEGF) Pathway
- VEGF-A (multiple isoforms: VEGF-A121, 165, 189, 206) is the primary driver
- VEGF-A binds VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk-1)
- VEGFR-2 signaling mediates: angiogenesis, vascular permeability, endothelial proliferation, migration
- In eye: VEGF-A overexpression drives choroidal neovascularization (CNV in AMD), retinal neovascularization (PDR), macular edema (BRVO/CRVO/DME)
- VEGF-B, VEGF-C, VEGF-D, PlGF also relevant (PlGF important in AMD)
Agents
Pegaptanib (Macugen)
- Type: RNA aptamer (binds and neutralizes VEGF-A165 isoform only)
- Dose: 0.3 mg intravitreal every 6 weeks
- Status: First FDA-approved anti-VEGF for wet AMD (2004); largely replaced by ranibizumab/bevacizumab
Ranibizumab (Lucentis)
- Type: Humanized monoclonal antibody Fab fragment (no Fc region)
- Target: All VEGF-A isoforms
- Dose: 0.5 mg/0.05 mL intravitreal monthly (or PRN/treat-and-extend)
- Half-life: ~9 days in vitreous; Fc-free design reduces systemic absorption
- FDA indications: Wet AMD, DME, BRVO/CRVO-associated macular edema, diabetic retinopathy, myopic CNV
- Clinical trials: MARINA, ANCHOR, RIDE/RISE, BRAVO/CRUISE, RESTORE
Bevacizumab (Avastin)
- Type: Full-length humanized IgG1 monoclonal antibody (Fc intact)
- Original indication: Colorectal cancer (IV)
- Ophthalmic use: Off-label (but widely used globally - cost-effective)
- Target: All VEGF-A isoforms
- Dose: 1.25 mg/0.05 mL intravitreal
- Half-life: Longer than ranibizumab (intact Fc → FcRn recycling)
- CATT Trial: Non-inferior to ranibizumab for visual acuity in wet AMD; significantly cheaper
- Compounding issue: Must be aseptically compounded from IV vials; contamination risk; shelf-life considerations
Aflibercept (Eylea, VEGF Trap)
- Type: Recombinant fusion protein - VEGFR-1 domain 2 + VEGFR-2 domain 3 fused to Fc of IgG1
- Targets: VEGF-A (all isoforms), VEGF-B, PlGF-1 and PlGF-2 (broader than ranibizumab/bevacizumab)
- Dose: 2 mg/0.05 mL; standard monthly × 3 then every 8 weeks; now high-dose 8 mg (Eylea HD/Eylea 8 mg) - q12-16 week dosing possible (PULSAR, PHOTON trials)
- Half-life in vitreous: ~9 days; VEGF-binding affinity significantly higher than ranibizumab
- FDA indications: Wet AMD, DME, BRVO/CRVO, diabetic retinopathy, ROP (retinopathy of prematurity - 2023 approval for ROP)
Faricimab (Vabysmo) - NEWEST FDA APPROVED (2022)
- Type: First bispecific antibody in ophthalmology; full IgG1 format
- Dual targets:
- VEGF-A (via ranibizumab-derived arm)
- Angiopoietin-2 (Ang-2) (via novel arm) - Ang-2 inhibition stabilizes vasculature, reduces inflammation and leakage (Tie-2 signaling pathway)
- Mechanism advantage: Ang-2 is upregulated in retinal disease and promotes VEGF-mediated destabilization; blocking both pathways simultaneously = synergistic stabilization
- Dose: 6 mg intravitreal; loading phase then q8-16 week personalized interval possible
- Clinical Trials:
- TENAYA/LUCERNE (wet AMD): Non-inferior to aflibercept; 45-46% achieved q16-week dosing
- YOSEMITE/RHINE (DME): Superior or non-inferior to aflibercept at 2 years; ~51-53% on q16-week dosing
- BALATON/COMINO (RVO): 2024 Phase 3 data - superior to ranibizumab at 24 weeks (PMID 38280653); 72-week treat-and-extend results published 2025 (PMID 40107501)
- MAGIC trial (Phase 2, non-proliferative DR): Ongoing (PMID 41587134)
- Source: TENAYA/LUCERNE anatomic outcomes, 2025
Brolucizumab (Beovu)
- Type: Single-chain antibody fragment (scFv) - smallest anti-VEGF biologic
- Target: All VEGF-A isoforms
- Advantage: Small size → higher molar concentration per injection; potentially longer durability (q12-week dosing)
- Serious adverse effect: Retinal vasculitis and retinal artery occlusion (rare ~3-4%; some cases severe vision loss) - requires careful monitoring; patients with prior ocular inflammation may be at higher risk
Conbercept (KH902) - Used in China
- Fusion protein similar to aflibercept; targets VEGF-A, VEGF-B, PlGF, VEGF-C
Recent Advances: Port Delivery System (PDS)
- Susvimo (ranibizumab PDS implant): Refillable intraocular implant surgically implanted in pars plana; delivers ranibizumab continuously; refilled every ~6 months; reduces injection burden; FDA approved 2021 (wet AMD), 2023 (DME); long-term trials (Archway) show non-inferiority to monthly injections
16. IMMUNOSUPPRESSANTS FOR DRY EYE
Cyclosporine A (Restasis 0.05%, Cequa 0.09%)
- Mechanism: Inhibits calcineurin → prevents dephosphorylation of NFAT → reduces T-cell activation and inflammatory cytokine (IL-2, IFN-γ, TNF) production in lacrimal gland → reduces T-lymphocyte mediated inflammation in ocular surface
- Increases goblet cell density, increases tear production by reducing lacrimal gland inflammation
- Pharmacokinetics: Topical; very low systemic absorption; nanoemulsion vehicle (Cequa 0.09% uses nanomicellar technology for better delivery); onset of clinical effect: 3-6 months
- Side effects: Burning/stinging (most common), redness
- Formulations: Restasis = 0.05% cationic nanoemulsion; Cequa = 0.09% nanomicellar
Lifitegrast (Xiidra 5%)
- Mechanism: LFA-1 (lymphocyte function-associated antigen-1, an integrin on T-cells) antagonist → blocks LFA-1 from binding ICAM-1 (intercellular adhesion molecule-1 on antigen-presenting cells) → inhibits T-cell activation and migration to ocular surface → reduces inflammation
- Novel mechanism: blocks the first step in T-cell activation at the ocular surface
- Pharmacokinetics: Topical BD; minimal systemic absorption
- Onset: Faster than cyclosporine (some benefit within 2 weeks for symptoms)
- Side effects: Instillation site discomfort, dysgeusia (altered taste - drug drains via nasolacrimal duct), transient visual disturbance
Topical Corticosteroids (Pulse therapy) for Dry Eye
- Short-term loteprednol, fluorometholone for acute flares; reduces inflammatory initiation cycle
17. SURGICAL ADJUNCTS IN OPHTHALMOLOGY
Antifibrotic Agents (Glaucoma Surgery Adjuncts)
- 5-Fluorouracil (5-FU): Pyrimidine analog; inhibits thymidylate synthase → inhibits DNA synthesis in fibroblasts → reduces subconjunctival scarring after trabeculectomy; given subconjunctivally intraoperatively or postoperatively (5 mg injections)
- Mitomycin C (MMC): Alkylating agent (crosslinks DNA); much more potent antifibrotic; single intraoperative application at trabeculectomy site (0.2-0.4 mg/mL for 1-5 minutes); also used in pterygium surgery (anti-recurrence), PRK (prevents haze), conjunctival/corneal tumors
- Risks: Thin avascular blebs → bleb leak, endophthalmitis; hypotony; limbal stem cell deficiency; corneal/scleral melt
Viscoelastics (Ophthalmic Viscosurgical Devices - OVDs)
- Sodium hyaluronate (Healon, Provisc): Highly viscous; maintains anterior chamber space, protects endothelium
- Hydroxypropylmethylcellulose (HPMC): Less viscous; less protection but easier removal
- Dispersive OVDs (Viscoat): Protect corneal endothelium better; used in phacoemulsification
- Cohesive OVDs (Healon GV): Better space maintenance; easier to remove
Intraocular Gases and Silicone Oil (Vitreoretinal Surgery)
- SF6 (20%): Lasts ~2 weeks; tamponade for retinal detachment
- C3F8 (14%): Lasts ~6-8 weeks; macular hole surgery
- Air: Shortest duration; pneumatic retinopexy
- Silicone oil: Permanent (must be removed surgically); complex retinal detachments; high risk proliferative vitreoretinopathy
Tissue Plasminogen Activator (tPA)
- Mechanism: Serine protease; converts plasminogen to plasmin → lyses fibrin clots
- Intravitreal use: Sub-retinal hemorrhage (submacular), intravitreal hemorrhage
- Intracameral use: Fibrin in anterior chamber post-surgery
Botulinum Toxin Type A (Botox)
- Mechanism: Cleaves SNAP-25 (synaptosomal-associated protein) → prevents ACh vesicle fusion → blocks neuromuscular transmission at extraocular muscles → temporary paralysis
- Ophthalmic uses: Strabismus treatment (weaken overacting muscle), blepharospasm, hemifacial spasm, entropion, cosmetic (periorbital wrinkles), thyroid eye disease (lid retraction)
- Duration: 3-4 months; repeat injections needed
18. DYES AND DIAGNOSTIC AGENTS
Fluorescein Sodium
- Type: Xanthene dye
- IV fluorescein angiography (FFA): 10-25% IV injection → rapid distribution to choroidal and retinal vessels → identifies CNV, ischemia, leakage; side effects include nausea/vomiting, skin yellowing (transient), anaphylaxis (rare, 1:2000)
- Topical: 0.25-2% strips or drops; stains denuded corneal epithelium (green with blue light); used for contact lens fitting, Goldman applanation tonometry (with 0.25% fluorescein + topical anesthetic)
Indocyanine Green (ICG)
- IV: Used for ICG angiography - penetrates through melanin/pigment → better visualization of choroidal circulation, polypoidal choroidal vasculopathy (PCV); binds plasma proteins
- Intravitreal: Vital dye for ILM (internal limiting membrane) staining during macular surgery (macular hole, ERM peeling)
- Side effects: Nausea, urticaria; contraindicated in iodine/shellfish allergy (contains iodine)
Brilliant Blue G (BBG) / Trypan Blue
- Intravitreal BBG: Stains ILM selectively (replacing ICG); safer profile
- Intracameral Trypan Blue 0.06%: Stains anterior capsule of lens for capsulorhexis in cataract surgery (especially white cataracts)
19. OCULAR TOXICITY OF SYSTEMIC DRUGS
| Drug | Ocular Effect | Notes |
|---|---|---|
| Chloroquine/Hydroxychloroquine | Bull's eye maculopathy (irreversible) - "chloroquine retinopathy"; cornea verticillata | Cumulative dose dependent; annual monitoring with HVF/SD-OCT/mfERG after 5 years |
| Ethambutol | Toxic optic neuropathy (bilateral central scotomas, color vision loss) | Dose and duration dependent; monthly color vision monitoring |
| Amiodarone | Cornea verticillata (benign, rarely affects vision); optic neuropathy (rare) | Deposits do not require drug stoppage |
| Tamoxifen | Macular crystalline deposits, CME, reduced visual acuity; PSC cataracts | Dose-dependent |
| Sildenafil/tadalafil | Bluish haze (PDE6 inhibition in rods); NAION risk (controversial) | Mild; reversible |
| Corticosteroids (systemic) | PSC cataracts, glaucoma, opportunistic infections, papilledema (on withdrawal = pseudotumor) | |
| Rifabutin | Uveitis/hypopyon (when combined with CYP3A4 inhibitors like clarithromycin) | Drug interaction |
| Isotretinoin | Dry eye, meibomian gland dysfunction, conjunctivitis | |
| Phenothiazines | Corneal/conjunctival/lens deposits; pigmentary retinopathy (thioridazine) | Thioridazine most toxic |
| Vigabatrin | Bilateral concentric visual field constriction (irreversible) | Regular perimetry monitoring required |
| Digitalis | Yellow-green color vision disturbance | Xanthopsia |
| Quinine | Retinal arteriolar spasm, cinchonism, blindness in overdose | |
| Dupilumab | Conjunctivitis, keratitis, blepharitis | Common (10-30%) in atopic dermatitis treatment |
20. PRESERVATIVES IN OPHTHALMIC FORMULATIONS
| Preservative | Mechanism | Concentration | Products | Toxicity |
|---|---|---|---|---|
| Benzalkonium chloride (BAC/BAK) | Quaternary ammonium; detergent - disrupts lipid membranes; denatures proteins | 0.004-0.02% | Most multi-dose drops (timolol, latanoprost, dorzolamide) | Most toxic; disrupts tear film lipid layer; corneal epithelial toxicity; conjunctival goblet cell loss; promotes allergic reactions; additive toxicity with prolonged use or multiple drops |
| Purite (stabilized oxychloro complex) | Breaks down to water and NaCl on contact with ocular surface | 0.005% | Alphagan P (brimonidine), Refresh Optive | Very low toxicity; "disappearing preservative" |
| SofZia (ionic buffered system) | Antimicrobial ionic system (borate buffer + zinc + sorbitol) | — | Travatan Z (travoprost) | Low toxicity; deactivated upon contact with ocular surface |
| Polyquaternium-1 (Polyquad) | Quaternary ammonium; larger molecule than BAK - penetrates less | 0.001% | Tobramycin-dexamethasone (some formulations) | Lower toxicity than BAK |
| Thimerosal | Organomercury; inhibits microbial SH enzymes | 0.005% | Older preparations (largely discontinued) | High allergy rate; removed from most modern formulations |
| Chlorhexidine | Disrupts bacterial membranes | 0.005-0.01% | Some preparations | Moderate ocular surface toxicity |
| EDTA (ethylene diamine tetraacetic acid) | Chelates divalent cations; synergistic with BAK | — | Co-preservative | Enhances corneal penetration when added with BAK |
| Phenylmercuric nitrate/acetate | Organomercury | — | Old formulations | Largely abandoned |
Preservative-Free (PF) Formulations
- Unit-dose vials (UDVs/minims): Single-use; no preservative needed; recommended for:
- Patients using >3 drops daily (high drop burden)
- Contact lens wearers
- Dry eye patients
- Pre/post-operative period
- Examples: Tafluprost PF (Saflutan), Preservative-free timolol, many lubricants
- A 2025 systematic review (PMID 41465776) confirmed that switching from preserved to PF prostaglandins significantly improves ocular surface parameters including corneal staining and TBUT
21. RECENT ADVANCES AND NOVEL DRUG DELIVERY
Gene Therapy
- Voretigene neparvovec (Luxturna): AAV2 vector delivering functional RPE65 gene; for RPE65-mutant Leber's congenital amaurosis / retinitis pigmentosa; subretinal injection; FDA approved 2017
- GT005 (Gyroscope): AAV-mediated complement factor I delivery for geographic atrophy - ongoing trials
- ADVM-022: Intravitreal AAV gene therapy delivering aflibercept gene construct; Phase 1/2 OPTIC trial ongoing
- 4D-150: Dual-target intravitreal gene therapy (VEGF-C + VEGF-A); Phase 2 PRISM trial for wet AMD
Port Delivery System
- Susvimo (ranibizumab implant): Surgical device implanted in pars plana; continuous drug release; refilled every 6 months; reduces injection burden
Sustained-Release Implants
- Ozurdex (dexamethasone 0.7 mg biodegradable PLGA implant): Approved; ~6 months
- Iluvien (fluocinolone acetonide 0.19 mg non-biodegradable): Approved; ~3 years
- Durysta (bimatoprost SR 10 µg biodegradable implant, intracameral): FDA approved 2020 for OAG; single biodegradable pellet placed in anterior chamber angle; releases bimatoprost for ~4 months; reduces injection frequency
Nanoparticle Drug Delivery
- Polymeric nanoparticles (PLGA, chitosan): Controlled release across ocular barriers; improved bioavailability
- Lipid nanoparticles (LNPs): Delivery of RNA therapeutics (siRNA, mRNA) to retina; active research area
- Mesoporous silica nanoparticles: Customizable surface chemistry; prolonged release (2026 review, PMID 42086969)
- Cyclodextrins: Complexing agents to solubilize lipophilic drugs in aqueous vehicles
- Contact lens drug delivery: Drug-impregnated contact lenses for sustained topical delivery
Micro/Nano Devices
- Microneedles: Minimally invasive delivery to suprachoroidal space or sclera; bypass subconjunctival barriers
- Suprachoroidal delivery: CLS-TA (triamcinolone acetonide suprachoroidal) - FDA approved 2021 (Xipere) for macular edema from uveitis
RNA Therapeutics
- Sirolimus (rapamycin): mTOR inhibitor; intravitreal for non-infectious uveitis (studies)
- Antisense oligonucleotides/siRNA: Experimental for AMD, DR, glaucoma neuroprotection
- Gene editing (CRISPR): Preclinical for dominant forms of RP
Emerging Antiglaucoma Targets
- Adenosine receptor agonists: A1 receptors in trabecular meshwork increase outflow
- Serotonin receptor modulators: 5-HT2A agonism increases uveoscleral outflow
- Neuroprotective agents: NMDA antagonists (memantine - failed Phase 3); CNTF (ciliary neurotrophic factor) - NT-501 implant (Renexus) for glaucoma neuroprotection under investigation
- Brimonidine neuroprotection: Independent of IOP lowering; activates Bcl-2 pathways, reduces RGC apoptosis
Dry Eye Advances
- Perfluorohexyloctane (PFHO) (Miebo 100%): FDA approved 2023 for dry eye associated with meibomian gland dysfunction; first drug specifically targeting evaporative dry eye; mechanism - semifluorinated alkane that reduces aqueous evaporation by stabilizing lipid layer; once-daily
- Varenicline nasal spray (Tyrvaya 0.03 mg): FDA approved 2021 for dry eye; intranasal delivery stimulates trigeminal-lacrimal reflex → increases basal tear production; mechanism: partial nicotinic acetylcholine receptor agonist on nasal mucosa
- REGN-A1H: Anti-Ang-2 + anti-VEGF for dry eye - experimental
SUMMARY: ANTIGLAUCOMA DRUG COMBINATIONS
| Combination | Brand | Components |
|---|---|---|
| Cosopt | Timolol 0.5% + Dorzolamide 2% | |
| Combigan | Timolol 0.5% + Brimonidine 0.2% | |
| Ganfort | Timolol 0.5% + Bimatoprost 0.03% | |
| DuoTrav | Timolol 0.5% + Travoprost 0.004% | |
| Xalacom | Timolol 0.5% + Latanoprost 0.005% | |
| Roclatan | Netarsudil 0.02% + Latanoprost 0.005% | Newest; works on both trabecular + uveoscleral |
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, Chapter 74 (Ophthalmology) - direct textbook reading
- Katzung's Basic and Clinical Pharmacology, 16th Ed
- Faricimab TENAYA/LUCERNE Phase 3: PMID 39617060
- Faricimab BALATON/COMINO RVO: PMID 38280653
- Faricimab YOSEMITE/RHINE epiretinal outcomes: PMID 40668667
- Preservative-free prostaglandins review: PMID 41465776
- Mesoporous nanoparticles for ocular delivery: PMID 42086969
- Dry eye advances & drug delivery systems: PMID 41994442
Explain the mechanism in detail
"aqueous humor" AND "intraocular pressure" AND "mechanism" AND "trabecular"
"ROCK inhibitor" AND "trabecular meshwork" AND "glaucoma" AND "mechanism"
"angiopoietin-2" AND "Tie2" AND "VEGF" AND "faricimab" AND "mechanism"
PMID: 37837578
PMID: 39708087
Complete Mechanisms of Action - Ophthalmic Drugs
FOUNDATIONAL PHYSIOLOGY: Aqueous Humor Dynamics
- Two-thirds formed by the ciliary body (non-pigmented ciliary epithelium) via active secretion - Na⁺/K⁺-ATPase actively pumps Na⁺ into the posterior chamber; bicarbonate (HCO₃⁻) and Cl⁻ follow passively; water follows osmotically. Carbonic anhydrase (CA-II and CA-XII isoforms) catalyzes CO₂ + H₂O → H₂CO₃ → H⁺ + HCO₃⁻, providing the bicarbonate for this transport.
- One-third by passive ultrafiltration from iris vessels
- Trabecular (conventional) pathway (~80-90%): Aqueous flows from posterior chamber → pupil → anterior chamber → trabecular meshwork (TM) → Schlemm's canal → collector channels → episcleral veins → systemic circulation. This is a pressure-dependent, flow-resistance pathway. The juxtacanalicular tissue (innermost TM layer, just adjacent to Schlemm's canal) provides the majority of outflow resistance.
- Uveoscleral (unconventional) pathway (~10-20%): Aqueous passes through the root of the iris and ciliary muscle spaces → supraciliary and suprachoroidal spaces → exits via scleral emissaria. This route is largely pressure-independent.
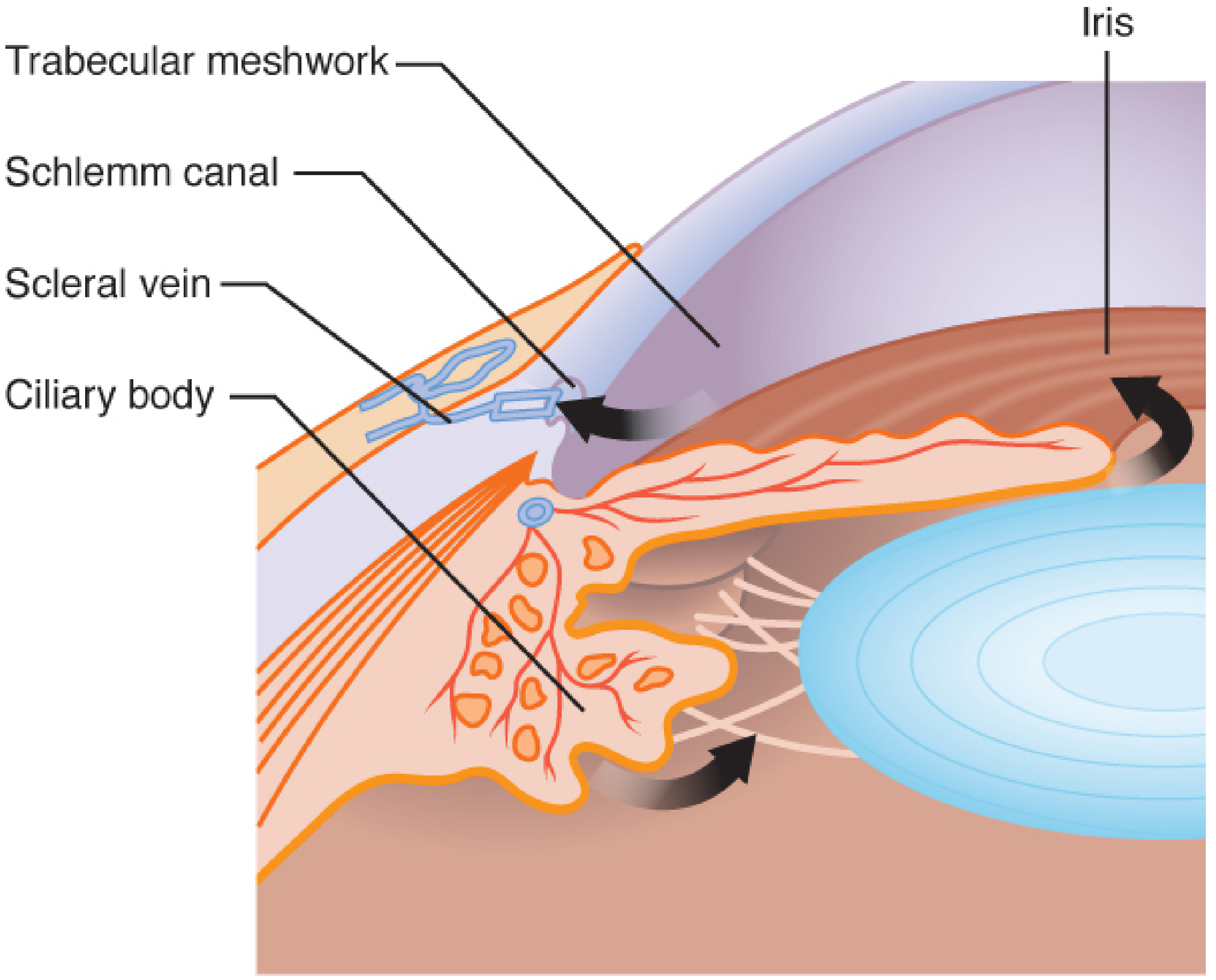
I. CHOLINERGIC (MUSCARINIC) AGONIST MECHANISMS
Pilocarpine - Step-by-Step Mechanism
- IP₃ (inositol 1,4,5-trisphosphate) → opens IP₃-gated Ca²⁺ channels on sarcoplasmic reticulum → [Ca²⁺]ᵢ rises
- DAG (diacylglycerol) → activates PKC (protein kinase C) → phosphorylates myosin light chain kinase (MLCK)
- Iris sphincter muscle (circumferential ring) contracts → pupil constricts
- In angle-closure glaucoma: miosis mechanically pulls iris away from trabecular meshwork → opens the drainage angle → reduces resistance → lowers IOP acutely
- Ciliary muscle is a ring-shaped muscle; contraction causes it to move anteriorly and inward
- This relaxes the zonular fibers → lens becomes more spherical → accommodation (near focus)
- More importantly for glaucoma: contraction pulls the scleral spur posteriorly and outward → mechanically opens the trabecular meshwork (widening inter-trabecular spaces) → increases conventional outflow → lowers IOP
- This is the primary mechanism in open-angle glaucoma
Pilocarpine
↓ binds M3 (Gq)
↓ PLC-β → IP3 + DAG
↓ IP3 → Ca²⁺ release
↓ Ca²⁺-CaM → MLCK activated
↓ Myosin phosphorylated
↓ Smooth muscle contraction
↙ ↘
Iris sphincter Ciliary muscle
contracts contracts
↓ ↓
Miosis Pulls scleral spur
(angle opens → TM opens
in ACG) → ↑ trabecular outflow
→ ↓ IOP (OAG)
Echothiophate - Indirect Mechanism (Anticholinesterase)
- Echothiophate phosphorylates the serine-OH in the esteratic active site (covalent bond)
- Forms an extremely stable phosphorylated AChE → irreversible inhibition (unlike carbamates, which are reversible)
- ACh accumulates at all cholinergic synapses → continuous muscarinic stimulation → sustained miosis + ciliary contraction → ↑ trabecular outflow → ↓ IOP
- Aging: With time (24-48 hrs), the phosphorylated enzyme undergoes "aging" (dealkylation) → becomes permanently refractory to reactivation by pralidoxime
- Recovery only occurs via synthesis of new AChE enzyme (weeks)
- Echothiophate also inhibits plasma cholinesterase (pseudocholinesterase)
- Succinylcholine is normally hydrolyzed by plasma cholinesterase in 3-5 minutes
- With echothiophate: succinylcholine half-life extends to 20-30 minutes → prolonged neuromuscular blockade → apnea → must discontinue 4-6 weeks before surgery using succinylcholine
II. MUSCARINIC ANTAGONIST MECHANISMS (MYDRIATICS/CYCLOPLEGICS)
- These drugs competitively bind M3 receptors on iris sphincter and ciliary muscle WITHOUT activating Gq signaling
- Block the binding of acetylcholine → prevent Gq → PLC-β → IP3 → Ca²⁺ pathway → NO contraction
- Result: Sympathetic tone is unopposed
- Iris sphincter paralyzed → iris dilator (alpha-1 adrenergic, sympathetically innervated) is unopposed → mydriasis
- Ciliary muscle relaxed → zonular fibers under tension → lens flattened → cycloplegia (paralysis of accommodation)
- TM tension reduced: With ciliary muscle relaxed, scleral spur is no longer pulled → TM inter-trabecular spaces may narrow slightly → slight outflow reduction (explains why these can precipitate angle closure)
- Atropine: very high affinity for M3, slow dissociation rate → 7-14 days
- Tropicamide: low affinity, rapid dissociation → 4-6 hours
- Cyclopentolate: intermediate affinity + significant blood-ocular barrier penetration → excellent cycloplegia for 12-24 hrs
- Mydriasis → lens-iris contact increases → aqueous cannot flow through pupil → pupillary block → aqueous pressure builds in posterior chamber → iris bulges forward (iris bombe) → peripheral iris occludes TM → acute angle closure glaucoma (medical emergency)
- Most dangerous in patients with shallow anterior chamber, thick peripheral iris, hypermetropes
III. BETA-ADRENERGIC BLOCKER MECHANISMS
Normal β-adrenergic signaling in ciliary body:
Norepinephrine/Epinephrine → β₂ receptor (Gs-coupled)
↓ Gαs activates adenylyl cyclase (AC)
↓ AC converts ATP → cAMP (↑ intracellular cAMP)
↓ cAMP activates PKA (protein kinase A)
↓ PKA phosphorylates ion channels and transport proteins
↓ Increases Na⁺/K⁺-ATPase activity + Cl⁻ secretion
↓ Water follows osmotically into posterior chamber
→ Aqueous humor PRODUCED
Beta-blocker mechanism:
Timolol (β₁ + β₂ blocker) → competitive antagonism at β₂ receptor on NPE
↓ Blocks Gαs activation → ↓ adenylyl cyclase
↓ ↓ cAMP → ↓ PKA activity
↓ ↓ Na⁺/K⁺-ATPase activity + ↓ Cl⁻ transport
↓ Less water secreted into posterior chamber
→ ↓ Aqueous humor PRODUCTION (~30-50%)
→ ↓ IOP (~20-30%)
- Ciliary body β-receptors are predominantly β₂ subtype
- Betaxolol's β₁ selectivity means it does not fully block the β₂ receptors driving aqueous production
- Net result: ~10-15% less IOP lowering than timolol
- But betaxolol has another effect: it blocks voltage-gated L-type Ca²⁺ channels in retinal ganglion cells → potential neuroprotective effect independent of IOP (may maintain retinal blood flow)
- Nasolacrimal drainage → systemic absorption → same β-blockade effects as oral beta-blockers
- β₁ blockade in heart → bradycardia, AV nodal depression, negative inotropy
- β₂ blockade in lungs → bronchospasm (particularly dangerous in asthma/COPD)
- β₂ blockade in pancreas → impairs glucagon-mediated glycogenolysis → hypoglycemia unawareness in diabetics
IV. ALPHA-2 ADRENERGIC AGONIST MECHANISMS
Mechanism 1 - Reduced Aqueous Production:
Brimonidine → α₂ receptor on NPE (Gi-coupled)
↓ Gαi INHIBITS adenylyl cyclase
↓ ↓ cAMP → ↓ PKA
↓ ↓ Na⁺/K⁺-ATPase → ↓ ion/water secretion
→ ↓ Aqueous production (~20-25%)
Mechanism 2 - Increased Uveoscleral Outflow:
α₂ receptor stimulation in ciliary body
↓ Activates Gi → but also activates other pathways
↓ Reduces ciliary muscle tone
↓ Opens supraciliary space
→ ↑ Uveoscleral outflow
Mechanism 3 - Neuroprotection (Independent of IOP):
Brimonidine crosses blood-retinal barrier
↓ α₂ receptor on retinal ganglion cells (RGCs)
↓ Gi → inhibits adenylyl cyclase
↓ Also activates PI3K/Akt (survival pathway)
↓ ↑ Bcl-2 (anti-apoptotic protein)
↓ ↓ Caspase-3 activation
→ ↓ RGC apoptosis
→ Neuroprotective effect independent of IOP lowering
V. PROSTAGLANDIN ANALOG MECHANISMS
FP Receptor Signaling Cascade:
Latanoprost (prodrug)
↓ Corneal esterases (in epithelium)
→ Latanoprost FREE ACID (active metabolite)
↓ Binds FP receptor (Gαq/11 + Gα12/13 coupled)
Gαq/11 → PLC-β → IP3 + DAG
IP3 → ↑ [Ca²⁺]ᵢ (from ER)
DAG → PKC activation
→ Ciliary muscle cells: contraction/relaxation changes
→ TM cells: altered contractility
Gα12/13 → activates RhoGEF (guanine nucleotide exchange factor)
→ RhoA-GTP (active RhoA)
→ Activates ROCK (Rho-associated coiled-coil forming kinase)
→ ROCK phosphorylates MLC (myosin light chain) and inhibits MLC phosphatase
(Note: in ciliary muscle, this leads to REMODELING rather than simple contraction)
Primary Mechanism: Uveoscleral Outflow Enhancement
FP receptor activation in ciliary muscle cells
↓
↑ Matrix metalloproteinase (MMP) expression:
MMP-1 (interstitial collagenase)
MMP-2 (gelatinase A)
MMP-3 (stromelysin)
MMP-9 (gelatinase B)
↓
Degradation of ECM components in ciliary muscle interstitial spaces:
Collagen types I, III, IV
Fibronectin
Laminin
↓
↓ Resistance to aqueous flow through ciliary muscle
↑ Aqueous percolation through supraciliary/suprachoroidal spaces
→ ↑ UVEOSCLERAL OUTFLOW (~40-100% increase)
→ ↓ IOP 25-35%
- Slight increase in trabecular outflow (less well characterized)
- Ciliary muscle contractile changes that widen the uveoscleral drainage angle
Why Once-Daily Evening Dosing?
- Peak uveoscleral outflow enhancement occurs 8-12 hours after dosing
- IOP peaks in early morning (06:00-09:00) due to cortisol surge and positional changes
- Evening dosing at ~21:00-22:00 → peak effect at ~06:00-09:00, precisely when IOP is highest
- The ECM remodeling effect also means some IOP reduction is maintained even when drug levels fall
Iris Pigmentation Mechanism:
FP receptors on iris melanocytes
↓ FP activation
→ ↑ Melanogenesis via Gαq → PKC pathway
→ ↑ Tyrosinase activity (rate-limiting enzyme in melanin synthesis)
→ ↑ Eumelanin production in iris melanocytes
→ Irreversible iris darkening (blue/green → brown)
Latanoprostene Bunod (LBN) - Novel Mechanism:
LBN in eye
↓ Hydrolysis
↓
FP receptor activation + NO released
(uveoscleral pathway) ↓
Activates soluble guanylyl cyclase (sGC)
↓ ↑ cGMP
↓ Activates PKG (cGMP-dependent protein kinase)
↓ Dephosphorylates myosin
↓ TM cell relaxation
↓ ↑ TRABECULAR outflow
VI. CARBONIC ANHYDRASE INHIBITOR MECHANISMS
Aqueous Production - Biochemical Basis:
CO₂ + H₂O ←—CA-II/CA-XII—→ H₂CO₃ → H⁺ + HCO₃⁻
HCO₃⁻ transported via NBC1 (Na⁺/HCO₃⁻ cotransporter) and AE2 (Cl⁻/HCO₃⁻ exchanger)
→ HCO₃⁻ accumulates on basolateral side of NPE
→ Na⁺ follows HCO₃⁻ via Na⁺/K⁺-ATPase
→ Osmotic water flux → aqueous humor formed
CAI inhibits CA-II (and CA-XII) in NPE
↓
↓ HCO₃⁻ production
↓
↓ Anion accumulation in NPE
↓
↓ Na⁺ transport (follows HCO₃⁻)
↓
↓ Osmotic water flux
↓
↓ Aqueous humor secretion (~30-50%)
↓
↓ IOP
- Dorzolamide/brinzolamide topically have low systemic absorption
- They distribute to red blood cells (high CA-II in RBCs) → long t½ in blood (~147 days for dorzolamide in RBCs)
- But low plasma concentrations mean low renal CA inhibition → fewer systemic side effects than oral acetazolamide
VII. RHO KINASE (ROCK) INHIBITOR MECHANISMS
Background - Rho/ROCK Pathway in Trabecular Meshwork:
Normal TM:
Rho GTPase (RhoA) is tonically active
→ ROCK phosphorylates:
(1) Myosin light chain (MLC) → actin-myosin contraction
(2) MYPT1 (MLC phosphatase target subunit) → INHIBITS MLC phosphatase
→ Net: TM cells are contracted/stiff
→ HIGH RESISTANCE to aqueous outflow
→ Contributes to elevated IOP
Netarsudil (prodrug) → corneal esterase → AR-13503 (active metabolite)
↓
Inhibits ROCK1 and ROCK2 (competitive ATP-site inhibitor)
↓
↓ Phosphorylation of MLC (less myosin activation)
↓ MYPT1 phosphorylation → MLC phosphatase ACTIVATED → dephosphorylates MLC
↓
↓ Actin stress fiber formation in TM cells
↓ Focal adhesion formation
↓ Cell stiffness (TM cells become more compliant)
↓
↑ Paracellular spaces in TM and Schlemm's canal inner wall
↑ Conventional (TRABECULAR) outflow facility
↓
↓ IOP ~20-25%
Mechanism 2 - NET (Norepinephrine Transporter) Inhibition:
AR-13503 also inhibits NET (norepinephrine reuptake transporter) at sympathetic nerve terminals
↓
↑ Norepinephrine remains in synaptic cleft
↑ α₂ receptor stimulation (via NE)
↓
Gi → ↓ cAMP in ciliary body epithelium
↓
↓ Aqueous humor production
Mechanism 3 - Decreased Episcleral Venous Pressure (EVP):
ROCK inhibition in episcleral veins
↓
Vasodilation of episcleral venous plexus
↓
↓ EVP (from ~8-10 mmHg normally)
↓
IOP = F/C + EVP → if EVP ↓, IOP ↓ directly
VIII. OSMOTIC AGENT MECHANISMS
IV Mannitol → distributed in plasma (does NOT cross into eye - large molecule)
↓
Creates osmotic gradient: [Plasma] > [Vitreous humor]
↓
Water moves from vitreous/aqueous → plasma (osmosis down the gradient)
↓
↓ Vitreous volume → ↓ IOP dramatically (within 30-60 min)
↓
Also: vitreous dehydration creates "negative pressure" → reduces lens forward pressure
- Acute angle closure glaucoma (shrinks vitreous → lens moves back → angle opens)
- Pre-surgical vitreous dehydration (creates space for anterior segment surgery)
IX. LOCAL ANESTHETIC MECHANISMS
Proparacaine/Tetracaine (tertiary amine; pKa ~9)
↓ At physiologic pH, both neutral + ionized forms exist
↓ NEUTRAL form crosses lipid membrane of nerve cell
↓ Once inside cell, equilibrium → ionized (NH⁺) form predominates
↓ Ionized form enters Nav channel pore from INSIDE (use-dependent block)
↓ Binds to local anesthetic receptor in channel pore
(segment S6 of domain IV in Nav1.7, Nav1.4)
↓ Physically occludes channel pore
↓ Channel cannot open → membrane cannot depolarize
↓ Action potential propagation BLOCKED in corneal sensory C and Aδ fibers
→ Anesthesia (loss of pain, touch, temperature)
- Loss of trophic support to corneal epithelium (neuropeptide substance P and CGRP from corneal nerves support epithelial cell renewal)
- Direct epithelial toxicity (detergent-like effect of high local concentrations)
- Impaired healing → neurotrophic keratopathy → corneal ulcer
X. ANTIBIOTIC MECHANISMS
1. Fluoroquinolones - DNA Gyrase & Topoisomerase IV Inhibition
Moxifloxacin/Ciprofloxacin enters bacterial cell
↓
GRAM-NEGATIVE primary target: DNA GYRASE (Topoisomerase II)
- Subunits: GyrA (2) + GyrB (2) = A₂B₂ tetramer
- Normal function: introduces negative supercoils ahead of replication fork
(relieves torsional stress during DNA replication/transcription)
↓
GRAM-POSITIVE primary target: TOPOISOMERASE IV (ParC + ParE subunits)
- Normal function: decatenation of daughter chromosomes after replication
↓
Drug-enzyme-DNA TERNARY COMPLEX forms:
Fluoroquinolone intercalates between the cut strands
+ Binds enzyme at the break point
↓
Creates "roadblock": replication forks collide with frozen enzyme-DNA complexes
↓
Double-strand DNA BREAKS accumulate
↓ (two bactericidal mechanisms)
(1) SOS response: recA-mediated → DNA degradation + irregular cell division
(2) Direct cell death from dsDNA breaks even without SOS
→ BACTERICIDAL
- Inhibit BOTH DNA gyrase AND topoisomerase IV with high affinity
- For resistance by mutation to emerge, BOTH enzyme targets must mutate simultaneously
- Mutation frequency: ~10⁻¹⁶ vs ~10⁻⁸ for 2nd-generation FQs → much lower resistance potential
2. Aminoglycosides - Ribosomal Misreading
Tobramycin (polycationic, basic drug)
↓
Electrostatic attraction to negative charge of LPS (lipopolysaccharide) on outer membrane
↓
Displaces Mg²⁺/Ca²⁺ (cross-bridges holding LPS together) → outer membrane disruption
↓ Initial uptake (oxygen-dependent, killed by anaerobes)
↓
Enters cytoplasm → binds 16S rRNA on 30S ribosomal subunit
(specifically the A-site on helix 44 of 16S rRNA)
↓
MISREADING mechanism:
Normal: cognate tRNA with matched anticodon → exact amino acid insertion
With aminoglycoside: drug distorts 16S rRNA A-site conformation
→ Near-cognate tRNAs (wrong amino acid) are accepted
→ MISINCORPORATION of amino acids → aberrant proteins
↓
Aberrant membrane proteins insert into cell membrane → INCREASED permeability
↓
More aminoglycoside enters → positive feedback → rapidly BACTERICIDAL
(Called the "self-promoted uptake" mechanism)
3. Chloramphenicol - Peptidyl Transferase Inhibition
Chloramphenicol → binds 23S rRNA on 50S ribosomal subunit
↓
Binds at the A-site of the PEPTIDYL TRANSFERASE CENTER (PTC)
↓
Blocks: aminoacyl-tRNA from entering the A-site
↓
Peptide chain CANNOT be elongated
→ BACTERIOSTATIC (does NOT kill; inhibits growth)
XI. ANTIVIRAL MECHANISMS
Acyclovir - Viral Selectivity Explained Step by Step
STEP 1 - ACTIVATION (key to selectivity):
Acyclovir (acycloguanosine) is a PRODRUG
↓
HSV-infected cells express viral THYMIDINE KINASE (TK)
↓
Viral TK phosphorylates acyclovir → acyclovir MONOPHOSPHATE (ACV-MP)
(Human TK does this ~1000x LESS efficiently → minimal toxicity to normal cells)
↓
Cellular kinases: ACV-MP → ACV-DP → ACV-TP (acyclovir triphosphate)
↓
STEP 2 - CHAIN TERMINATION:
ACV-TP competes with dGTP (deoxy-guanosine triphosphate) for viral DNA polymerase
↓
ACV-TP has ~100-fold higher affinity for viral DNA pol than human DNA pol
↓
ACV-TP incorporated into growing viral DNA chain (in place of dGMP)
↓
PROBLEM: acyclovir lacks the 3'-OH group of normal nucleosides
(acyclic side chain = no ring, no 3'-OH)
↓
DNA chain TERMINATES (no 3'-OH means next nucleotide cannot be added)
↓
ADDITIONALLY: viral DNA polymerase becomes irreversibly TRAPPED on the chain
(suicidal enzyme inactivation)
→ Viral DNA replication completely halted
→ VIROSTATIC (prevents viral replication; does NOT kill existing virus)
- TK mutation: virus loses TK → cannot phosphorylate acyclovir → resistant (TK-deficient mutants)
- DNA pol mutation: altered polymerase has lower affinity for ACV-TP
- TK-negative mutants are less virulent (TK needed for efficient neuronal reactivation) but dangerous in immunocompromised patients
- Treatment of resistant HSV: Foscarnet (does NOT need TK activation - directly inhibits DNA pol) or cidofovir
Ganciclovir - Same but for CMV
Ganciclovir (GCV) in CMV-infected cells
↓
CMV UL97 kinase (NOT TK - different kinase) phosphorylates GCV → GCV-MP
↓
Cellular kinases → GCV-TP
↓
Inhibits CMV DNA polymerase (UL54)
→ Chain termination (like acyclovir)
→ CMV DNA replication inhibited
Foscarnet (phosphonoformate)
↓
DIRECTLY inhibits viral DNA polymerase at the PYROPHOSPHATE BINDING SITE
(blocks pyrophosphate release during nucleotide incorporation)
↓
Inhibits: HSV DNA pol, CMV DNA pol (UL54), HIV reverse transcriptase
↓
Does NOT need phosphorylation → effective even against TK-deficient (acyclovir-resistant) HSV
XII. ANTIFUNGAL MECHANISMS
Natamycin (Polyene) - Ergosterol Binding
Natamycin molecule contains a large lactone ring with alternating conjugated double bonds
↓
Binds ERGOSTEROL (primary sterol in fungal cell membranes)
(Humans use CHOLESTEROL; this difference provides selectivity)
↓
Drug-ergosterol complex inserts into the membrane
↓
Forms PORES/CHANNELS (not a specific pore structure - general membrane disruption)
↓
K⁺ leaks out, small cations and molecules leak in
↓
Membrane potential collapses → cell contents leak out → FUNGICIDAL
Azoles (Voriconazole) - Ergosterol Synthesis Inhibition
Voriconazole enters fungal cell
↓
Inhibits CYP51 (lanosterol 14α-demethylase) - a fungal cytochrome P450 enzyme
↓
BLOCKS conversion: Lanosterol → Eburicol → (several steps) → Ergosterol
↓
Ergosterol NOT produced
↓
(1) Membrane loses fluidity (ergosterol maintains membrane function)
(2) Toxic methylated sterols (14α-methyl sterols) ACCUMULATE
→ Inhibit membrane-bound enzymes
→ Cell growth inhibited (FUNGISTATIC) or death (FUNGICIDAL for some)
XIII. CORTICOSTEROID MECHANISMS
At the Molecular Level:
Prednisolone acetate (lipophilic) penetrates cell membrane
↓
Binds cytoplasmic GLUCOCORTICOID RECEPTOR α (GRα) - a ligand-activated transcription factor
↓
Drug-GR complex dissociates from HSP90 (heat shock protein 90) chaperone
↓
GR undergoes conformational change → nuclear localization signals exposed
↓
Drug-GR complex translocates to NUCLEUS
↓
Dimerizes and binds GLUCOCORTICOID RESPONSE ELEMENTS (GREs) in DNA
1. TRANSACTIVATION (GRE-binding):
GR-GRE binding → ↑ transcription of:
- Lipocortin 1 (Annexin A1): inhibits phospholipase A2 → ↓ arachidonic acid release
- MAPK phosphatase-1 (MKP-1): inactivates ERK/JNK/p38 MAPK cascades
- IκBα: inhibits NF-κB nuclear entry
- IL-10: anti-inflammatory cytokine
2. TRANSREPRESSION (protein-protein interaction without DNA binding):
GR monomer directly interacts with:
- NF-κB: blocks transcription of TNF-α, IL-1, IL-6, IL-8, COX-2, iNOS
- AP-1 (Fos/Jun dimer): blocks matrix metalloproteinase production
3. NON-GENOMIC (rapid, within minutes):
- Direct membrane effects on eicosanoid synthesis
- Annexin-1 release → rapid PLA2 inhibition
- Vasoconstrictive effect on conjunctival/corneal blood vessels (reduces redness)
- ↓ Prostaglandins (PLA2 inhibition) → ↓ vascular permeability, ↓ chemotaxis
- ↓ Cytokines (TNF-α, IL-1, IL-6) → ↓ cellular infiltration
- ↓ Histamine release from mast cells
- Stabilizes lysosomal membranes → ↓ tissue-damaging enzyme release
Steroid Glaucoma Mechanism:
Glucocorticoids in TM cells
↓ GR activation
↓ ↑ Myocilin (MYOC) expression
↓ Myocilin accumulates in TM ECM → glycosaminoglycan accumulation
↓ TM cells become stiffer (increased actin stress fibers, crosslinked ECM)
↓ ↓ Phagocytic activity of TM cells
↓ ↓ Conventional outflow facility
↓ ↑ IOP
→ STEROID-INDUCED OCULAR HYPERTENSION (SIOH) in ~30% of general population
("Steroid responders" have GRα polymorphisms with higher TM sensitivity)
Loteprednol - "Retro-metabolic" Design:
Loteprednol binds GR → anti-inflammatory effect (same genomic mechanism)
↓
After receptor binding, loteprednol undergoes PREDICTABLE OXIDATIVE METABOLISM
→ Converts to inactive metabolite Δ1-cortienic acid etabonate
↓
Inactive metabolite has NO glucocorticoid activity
→ ↓ Duration of action → ↓ cumulative steroid load in TM/lens
→ ↓ IOP elevation risk
→ ↓ PSC cataract risk
XIV. NSAID MECHANISMS
COX Inhibition Cascade:
Injury/inflammation → phospholipid membrane disruption
↓ Phospholipase A2 (PLA2)
→ ARACHIDONIC ACID released
↓ Cyclooxygenase-1 (COX-1) or COX-2
→ PGG2 (prostaglandin G2)
↓ Peroxidase activity of COX
→ PGH2 (prostaglandin H2)
↓
Tissue-specific synthases:
PGH2 → PGE2 (by PGES) → pain, vasodilation, fever, hyperalgesia
PGH2 → PGI2 (by PGIS) → vasodilation, inhibit platelet aggregation
PGH2 → TXA2 (by TXAS) → vasoconstriction, platelet aggregation
Ketorolac/Diclofenac/Bromfenac
↓
Compete with arachidonic acid for the COX active site
(fits into the hydrophobic channel of COX)
↓
↓ Prostanoid synthesis
↓
In eye: ↓ PGE2 → ↓ vascular permeability → ↓ CME
↓ PGI2 → ↓ vasodilation
↓ TXA2 → less effect on platelet aggregation (topical)
↓ Intraoperative PG release from iris trauma → prevents surgically-induced miosis
(PGE2 normally released during surgical manipulation causes miosis via EP2/EP4 receptors)
Nepafenac → corneal amidases → AMFENAC (active NSAID)
↓
Amfenac penetrates corneal stroma and anterior chamber efficiently
→ Reaches the ciliary body and retina at higher concentrations
→ Better intraocular bioavailability vs diclofenac/ketorolac
XV. ANTI-VEGF BIOLOGIC MECHANISMS
VEGF Signaling Pathway (What these drugs block):
VEGF-A (165 isoform most important)
↓
Binds VEGFR-2 (KDR/Flk-1) → receptor DIMERIZES
↓
Dimerization → transphosphorylation of intracellular tyrosine kinase domains
↓
Phosphotyrosines recruit adaptor proteins:
- PLCγ → IP3/DAG → Ca²⁺/PKC → proliferation, migration
- PI3K → Akt/PKB → cell SURVIVAL, migration, VEGFR endocytosis
- Ras/MAPK → ERK1/2 → proliferation, VEGF production (positive feedback)
- eNOS phosphorylation → ↑ NO → vasodilation, ↑ vascular permeability
- Src kinase → disrupts VE-cadherin at endothelial junctions → ↑ permeability
↓
NET EFFECTS in retinal pathology:
↑ Vascular permeability (endothelial junction disruption)
↑ Neovascularization (endothelial cell proliferation/migration)
↑ Macular edema (fluid accumulation in retinal layers)
Drug-Target Interactions:
- Binds all VEGF-A isoforms at the receptor-binding domain
- The Fab fragment (no Fc region) → CANNOT be recycled by FcRn → shorter vitreous half-life
- Does NOT bind VEGF-B, PlGF
- Same anti-VEGF-A epitope as ranibizumab (derived from same parent mouse antibody)
- Intact Fc → FcRn-mediated recycling → longer systemic half-life (concern for systemic anti-VEGF effects with intravitreal use via transscleral absorption)
Structure: VEGFR-1 D2 + VEGFR-2 D3 + IgG1 Fc
VEGFR-1 domain 2 binds: VEGF-A (all isoforms) + PlGF-1 + PlGF-2
VEGFR-2 domain 3 binds: VEGF-A (with very high affinity) + VEGF-B
IgG1 Fc: FcRn recycling (extends half-life in vitreous)
- Binding affinity for VEGF-A: Kd ~1 fM (vs ~60 fM for ranibizumab)
- ~100-fold higher affinity for VEGF-A than native receptors → acts as a "VEGF sink"
- PlGF blockade: important because PlGF is elevated in AMD and promotes macrophage-driven CNV
- High-dose 8 mg (Eylea HD): higher molar dose → longer duration of effect (q12-16 week dosing possible)
Faricimab - Bispecific Dual Pathway Block:
VEGF-A arm (ranibizumab-derived):
Blocks VEGF-A → VEGFR-2 signaling (as above)
→ ↓ Neovascularization, ↓ vascular permeability
ANGIOPOIETIN-2 (Ang-2) arm:
Background: Ang-1 binds Tie-2 receptor → STABILIZES vasculature
(phosphorylates Tie2 → PI3K/Akt → promotes pericyte attachment,
tight junctions, endothelial survival)
In disease: Ang-2 is UPREGULATED (released from Weibel-Palade bodies under stress)
Ang-2 COMPETES with Ang-1 for Tie2 → BLOCKS Tie2 signaling
→ Pericyte dropout, tight junction disruption, inflammation, fibrosis
Faricimab → NEUTRALIZES Ang-2
↓
Ang-1/Tie2 signaling RESTORED (unopposed)
↓
PI3K → Akt activation in endothelial cells:
↑ VE-cadherin at junctions → tighter junctions → ↓ permeability
↑ Pericyte recruitment → vascular stability
↑ eNOS → anti-inflammatory signaling
↓ NF-κB → ↓ ICAM-1, ↓ VCAM-1 → ↓ leukostasis/inflammation
↓ Ang-2 activates integrin-αvβ3/αvβ5 → promotes ERM/fibrosis - BLOCKED
SYNERGY:
VEGF-A blockade ↓ neovascularization (angiogenic drive)
Ang-2 blockade ↓ vascular instability (stability pathway restored)
Together: ↓ fluid, ↓ neovascularization, ↓ inflammation, ↓ fibrosis
→ More complete and durable vascular stabilization than anti-VEGF monotherapy
XVI. IMMUNOSUPPRESSANT MECHANISMS (DRY EYE)
Cyclosporine A - Calcineurin Inhibition:
Cyclosporine enters T-lymphocyte (especially CD4+ Th1 cells)
↓
Binds CYCLOPHILIN (cytoplasmic peptidyl-prolyl isomerase)
↓
CyA-Cyclophilin complex binds CALCINEURIN (Ca²⁺/CaM-dependent phosphatase)
↓
Calcineurin INHIBITED → cannot dephosphorylate NFAT (nuclear factor of activated T cells)
↓
NFAT-P (phosphorylated) cannot translocate to nucleus
↓
No NFAT binding to IL-2 gene promoter
↓
↓ IL-2 transcription → ↓ IL-2 production
↓
Without IL-2 autocrine signal, T cells CANNOT proliferate
↓
Downstream: ↓ IFN-γ, ↓ TNF-α, ↓ IL-1β production
↓
↓ Lacrimal gland inflammation
↓ Conjunctival T-cell density
↑ Goblet cell density (inflammatory suppression allows recovery)
↑ Tear production (lacrimal gland function restored)
Lifitegrast - LFA-1/ICAM-1 Blockade:
Dry eye inflammatory cycle:
Environmental stress → ↑ ICAM-1 on ocular surface epithelium
↓
ICAM-1 binds LFA-1 (lymphocyte function-associated antigen-1, an integrin α_L β_2)
on T-lymphocyte surface
↓
LFA-1/ICAM-1 interaction activates T cell:
(1) Provides co-stimulatory signal
(2) Facilitates T cell migration and adhesion to ocular surface
↓
T cells activated → ↑ IL-1β, ↑ MMP-3, ↑ MMP-9 → damage mucins, goblet cells
↓ Tear stability → ↑ osmolarity → ↑ stress → MORE ICAM-1 (vicious cycle)
Lifitegrast blocks:
LFA-1 (binds to α_L subunit I-domain - same site as ICAM-1 but non-competitive
- it occupies the binding groove, preventing ICAM-1 from inserting)
↓
No LFA-1/ICAM-1 ligation → T cell NOT co-stimulated
↓
↓ T cell activation, ↓ T cell migration to ocular surface
↓
↓ IL-1β, ↓ TNF-α, ↓ MMPs → ↓ inflammation → ↓ dry eye symptoms
XVII. BOTULINUM TOXIN MECHANISM
Botulinum toxin type A (BoNT-A) is a ~150 kDa zinc metalloprotease
↓
STEP 1 - BINDING:
Heavy chain (HC, C-terminal) binds polysialoganglioside receptors (GT1b, GD1a)
+ SV2C (synaptic vesicle protein 2C) at presynaptic cholinergic nerve terminals
↓
STEP 2 - ENDOCYTOSIS:
Receptor-mediated endocytosis → BoNT-A enters acidic endosome
↓
STEP 3 - TRANSLOCATION:
Acid-induced conformational change → light chain (LC) translocates across
endosomal membrane into cytosol (pore-forming mechanism of HC N-terminal domain)
↓
STEP 4 - PROTEOLYSIS:
LC is a zinc-dependent endopeptidase
BoNT-A specifically cleaves SNAP-25 (synaptosomal-associated protein 25 kDa)
at a single Gln197-Arg198 peptide bond
↓
SNAP-25 is a component of the SNARE complex:
(SNARE = Soluble NSF Attachment protein Receptor)
VAMP/synaptobrevin (on vesicle) + SNAP-25 (on plasma membrane) + Syntaxin-1 (on PM)
form the SNARE complex → drives membrane fusion → ACh vesicle exocytosis
↓
With SNAP-25 cleaved → SNARE complex CANNOT form → NO vesicle fusion
↓
No ACh released at neuromuscular junction → muscle CANNOT contract
↓ (at extraocular muscles)
Muscle weakened/paralyzed
↓
REVERSIBILITY: New SNAP-25 synthesized over 3-4 months → function returns gradually
- Strabismus: Injected into overacting rectus muscle → temporary paralysis → allows antagonist to tighten → long-term alignment correction (even after toxin wears off)
- Blepharospasm: Injected into orbicularis oculi → ↓ spasm → improved eye opening
- Lid retraction in thyroid eye disease: ↓ superior tarsal muscle (Müller's) and levator action
XVIII. ANTIALLERGIC MECHANISM - MAST CELL PATHWAY
Complete allergic conjunctivitis cascade:
SENSITIZATION:
Allergen → corneal/conjunctival antigen-presenting cells (APCs)
↓ Process and present to CD4+ T cells (Th2 polarization)
↓ IL-4, IL-13 production by Th2 cells
↓ B cell class switch → IgE production
↓ IgE binds FcεRI (high-affinity IgE receptor) on MAST CELLS
(conjunctival mast cells: ~50 million/eye; palpebral > bulbar)
RE-EXPOSURE (EARLY PHASE, seconds-minutes):
Allergen cross-links IgE-FcεRI complexes on mast cell surface
↓
FcεRI receptor aggregation → activation of:
(1) Lyn kinase (Src family) → phosphorylates ITAMs on FcεRI
(2) Syk kinase → recruited and activated
(3) LAT scaffold phosphorylated → recruits PLCγ
↓
PLCγ → IP3 + DAG
IP3 → ER Ca²⁺ release + CRAC channel opening (SOCE) → ↑ [Ca²⁺]ᵢ
DAG → PKC activation
↓
Ca²⁺-dependent DEGRANULATION:
Preformed mediators RELEASED:
- HISTAMINE → H1 receptors on conjunctival vessels + sensory nerves
→ vasodilation, ↑ permeability (edema/chemosis), pruritus
- Tryptase (marker of mast cell degranulation)
- Heparin, proteoglycans
LATE PHASE (2-4 hours):
Newly synthesized mediators (from arachidonic acid):
- PGD2 → DP2 receptor → Th2 chemotaxis
- LTC4/D4/E4 (cysteinyl leukotrienes) → further permeability, chemotaxis
- PAF → eosinophil recruitment
↓
Eosinophil infiltration → eosinophil cationic protein (ECP), MBP → corneal damage (VKC)
-
Cromolyn/Lodoxamide (mast cell stabilizers): Inhibit Cl⁻ channel opening that normally follows Ca²⁺ influx during mast cell activation → prevents degranulation. Must be used BEFORE allergen exposure (prophylactic only).
-
H1 antihistamines (olopatadine, ketotifen): Competitive antagonists at H1 receptor on:
- Conjunctival vessels → block histamine-mediated vasodilation/permeability
- Sensory nerve endings → block histamine-mediated itch (pruritus via TRPV1/TRPA1 potentiation by H1 signaling on C fibers)
-
Dual agents (olopatadine): BOTH block H1 AND prevent mast cell degranulation (blocks calcium influx) → cover both the symptom AND the release.
-
Alcaftadine (unique): Also blocks H2 receptors (which mediate pruritus via different pathway) + prevents eosinophil transmigration → useful in severe VKC.
MECHANISM SUMMARY TABLE
| Drug Class | Primary Receptor/Target | Second Messenger | Net Effect on IOP/Inflammation |
|---|---|---|---|
| Pilocarpine | M3 (Gq) | IP3/Ca²⁺/PKC | ↑ TM outflow via scleral spur traction |
| Timolol | β₂ (Gi to AC) | ↓ cAMP | ↓ Aqueous production (30-50%) |
| Brimonidine | α₂ (Gi) | ↓ cAMP | ↓ Production + ↑ uveoscleral outflow |
| Latanoprost | FP (Gαq + Gα12/13) | IP3/Ca²⁺ + MMP upregulation | ↑ Uveoscleral outflow via ECM remodeling |
| Dorzolamide | CA-II enzyme | ↓ HCO₃⁻ | ↓ Aqueous secretion (ion transport) |
| Netarsudil | ROCK1/2 + NET | ↓ Actin stress fibers | ↑ TM outflow + ↓ production + ↓ EVP |
| Prednisolone | GRα (nuclear) | GRE transactivation/transrepression | ↓ NF-κB, ↓ PLA2, ↓ all inflammation |
| Acyclovir | Viral TK + DNA pol | Chain termination | Blocks HSV DNA replication |
| Ranibizumab | VEGF-A (direct binding) | No 2nd messenger - neutralization | ↓ VEGFR-2 signaling, ↓ permeability/CNV |
| Faricimab | VEGF-A + Ang-2 (bispecific) | Restores Tie2/Akt + blocks VEGFR-2 | ↓ Permeability + ↑ vascular stability |
| Cyclosporine | Cyclophilin → Calcineurin | ↓ NFAT dephosphorylation → ↓ IL-2 | ↓ T-cell activation → ↓ dry eye |
| Lifitegrast | LFA-1 I-domain | Blocks ICAM-1 binding | ↓ T-cell co-stimulation → ↓ ocular inflammation |
| Botulinum A | SNAP-25 (SNARE protease) | SNARE disruption | No ACh release → muscle paralysis |
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, 14th Ed, Chapter 74 (direct reading)
- Miller's Anesthesia, 10th Ed (aqueous humor physiology, IOP anatomy)
- ROCK inhibitors mechanism review: PMID 37837578
- Faricimab Ang-2/VEGF dual mechanism: PMID 39708087
- Glucocorticoid-induced IOP mechanism: PMID 38379915
Is this enough for my viva and theory
What You Have - Strengths
What Might Still Be Asked - Gaps to Address
1. Specific Dosing / Preparation Details
- "What is the concentration of pilocarpine used in glaucoma?" (1-4%)
- "What is the dose of intravitreal bevacizumab?" (1.25 mg/0.05 mL)
- "What are the contents of Tobradex?" (tobramycin 0.3% + dexamethasone 0.1%)
2. Drug Interactions Table
- Brimonidine + MAO inhibitors (dangerous hypertensive/hypotensive crisis)
- Topical beta-blockers + systemic beta-blockers (additive cardiovascular depression)
- Fluoroquinolones + antacids/metal cations (chelation - less relevant for ophthalmic drops but asked in theory)
- Cyclosporine + CYP3A4 inhibitors (azoles raise systemic cyclosporine levels)
3. Specific Conditions → Drug of Choice
4. Fluorescein Staining Patterns
- Rose Bengal: stains dead/devitalized epithelial cells (not covered in detail)
- Lissamine green: similar to rose Bengal, better tolerated
- Fluorescein: stains denuded epithelium (covered)
- These are short 2-mark questions but easy marks
5. Short Notes Commonly Asked in Theory
- Ocusert (pilocarpine sustained release insert)
- Durysta (intracameral bimatoprost implant)
- Miotics in glaucoma (with a diagram of TM mechanism)
- Steroid responders (mechanism of steroid glaucoma)
- Preservative toxicity of BAK (already covered well)
- Topical anesthetic abuse (neurotrophic keratopathy mechanism - covered)
6. What Was NOT Covered At All
- Subconjunctival/retrobulbar injection technique and drugs (lignocaine + hyaluronidase for peribulbar block)
- Hyaluronidase used in periocular blocks (breaks hyaluronic acid in tissues → improves spread of LA)
- Viscoelastics in more detail (cohesive vs dispersive classification for exam)
- Enzymatic vitreolysis - Ocriplasmin (jetrea): serine protease that cleaves fibronectin and laminin at vitreoretinal interface for vitreomacular traction
Honest Bottom Line
| Exam Type | Coverage Assessment |
|---|---|
| MBBS Final theory | 95% covered - you can write full answers on any topic |
| MBBS Viva | 90% - add drug-of-choice quick recall |
| MD/DNB Pharmacology theory | 85% - add drug interactions table + dosing numbers |
| MD/DNB Ophthalmology viva | 80% - add surgical drugs (viscoelastics, gases, ocriplasmin) in more depth |
| Pharm.D / Clinical pharmacy | 90% - mechanisms are very strong; add monitoring parameters |
MD
"ophthalmic drug" AND "clinical pharmacology" AND "glaucoma" AND "review"
"intravitreal injection" AND "pharmacokinetics" AND "anti-VEGF" AND "vitreous"
"neuroprotection" AND "glaucoma" AND "retinal ganglion cell" AND "drug"
"dry eye" AND "treatment" AND "perfluorohexyloctane" OR "varenicline" AND "ophthalmic"
"hydroxychloroquine" AND "retinal toxicity" AND "screening" AND "guidelines"
"trabecular meshwork" AND "emerging" AND "glaucoma" AND "drug target"
MD Ophthalmology - Honest Assessment & What You Still Need
Straight Answer First
What MD Ophthalmology Examiners Actually Ask (Based on Pattern Analysis)
Category 1: "Explain your choice of drug in this clinical scenario"
Category 2: Surgical pharmacology (a major gap in what was covered)
Category 3: Drug monitoring protocols with specific thresholds
Category 4: Emerging/pipeline drugs (expected at MD level)
Category 5: Pharmacology of specific diseases as a whole (not just drug classes)
SUPPLEMENTARY CONTENT FOR MD OPHTHALMOLOGY
A. VISCOELASTICS - OPHTHALMIC VISCOSURGICAL DEVICES (OVDs)
Classification by Rheological Properties
- Cohesive OVDs: High molecular weight, high viscosity, high elasticity. Molecules stay together (cohesive). Maintain space well. Easy to remove in one piece at end of surgery.
- Dispersive OVDs: Lower molecular weight, lower viscosity, low surface tension. Coat surfaces. Do NOT maintain space as well. Harder to completely remove (fragments remain).
| Property | Cohesive | Dispersive |
|---|---|---|
| Viscosity | Very high | Moderate-low |
| Space maintenance | Excellent | Poor |
| Endothelial protection | Moderate | Excellent (coat surfaces) |
| Ease of removal | Easy | Difficult (leaves fragments) |
| Risk if retained | IOP spike | Prolonged but less severe IOP rise |
| Best use in phaco | Creates space for capsulorhexis, IOL implantation | Protects corneal endothelium during ultrasound |
Specific Agents
- Glycosaminoglycan; negatively charged; binds water (holds 1000x its weight in water)
- Healon (1% HA, MW ~4 million Da): Cohesive; excellent space maintenance
- Healon GV (1.4% HA, MW ~5 million Da): Even more cohesive; "super cohesive"
- Healon 5 (2.3% HA): Viscoadaptive - behaves cohesively at low flow (maintains space) and dispersively at high phaco power (protective coating) → "viscoadaptive" or "viscous dispersive" behavior
- Provisc (1% HA): Similar to Healon; from Alcon
- Viscoat (4% HA + 3% chondroitin sulfate): Dispersive; best endothelial protection during phaco
- Sulfated glycosaminoglycan; negatively charged; lubricative
- Used in combination with HA (Viscoat); CS alone is low molecular weight → dispersive behavior
- Semi-synthetic polymer; 2% HPMC
- Much cheaper than HA-based OVDs
- Intermediate viscosity; less cohesive than Healon
- Widely used in low-resource settings; adequate for routine cataract surgery
- Degrades slower than HA → slightly higher risk of post-op IOP spike
- Orcolon: rarely used now
"Soft Shell" Technique (Arshinoff):
- Inject Viscoat (dispersive) first - fills posterior chamber and coats endothelium
- Inject Healon (cohesive) on top - pushes Viscoat to periphery and back, maintains working space centrally
- After phaco: Cohesive Healon removed easily first; Viscoat (which protected endothelium throughout) removed last
- This gives maximum endothelial protection (dispersive) PLUS maximum space maintenance (cohesive)
Post-Operative IOP Spike Mechanism:
- Cohesive OVDs: large molecules → TM physically blocked → larger IOP spike but shorter duration (cleared faster)
- Dispersive OVDs: smaller, stickier → TM coated with thin layer → smaller spike but more prolonged
- Prevention: thorough removal at end of surgery; brimonidine drops post-op
B. OCULAR ANESTHESIA FOR SURGERY - COMPLETE
Topical Anesthesia (for cataract surgery)
- Proparacaine 0.5% + intracameral lidocaine 1% (preservative-free, 0.5 mL)
- Safest; no risk of retrobulbar hemorrhage, globe perforation, brainstem anesthesia
- Requires cooperative patient; no akinesia
Peribulbar Block (preferred over retrobulbar now)
- Lignocaine (lidocaine) 2%: Onset 2-5 min, duration 1-2 hrs; amide LA
- Bupivacaine 0.5%: Onset 10-15 min, duration 4-6 hrs; amide LA; for longer procedures
- Mixture: Lignocaine 2% + Bupivacaine 0.5% (rapid onset of lignocaine + long duration of bupivacaine)
- Hyaluronidase 3-7.5 IU/mL: Added to LA mixture
- Mechanism: Depolymerizes hyaluronic acid in the orbital connective tissue matrix (hydrolyzes β-1,4 glycosidic bonds) → breaks down "tissue cement" → improves spread of LA through orbital fat → better akinesia with smaller volume, less orbital pressure, less risk of globe perforation
- Does NOT prolong anesthesia; purely enhances spread
- Epinephrine 1:100,000 (sometimes): Vasoconstriction → slows LA absorption → prolongs duration + reduces systemic toxicity
Retrobulbar Block (now less preferred)
- Drug: Lignocaine 2% ± bupivacaine, with hyaluronidase
- Injected behind the globe, within the muscle cone
- Better akinesia, less volume needed
- Higher risk: retrobulbar hemorrhage, globe perforation (esp myopic eyes), optic nerve damage, brainstem anesthesia (via spread along optic nerve sheath), oculocardiac reflex
Sub-Tenon's Anesthesia
- Drug: Lignocaine 2% (2-5 mL)
- Cannula inserted through small conjunctival incision into sub-Tenon's space
- No needle near orbit → safer; good for posterior segment surgery
- Provides analgesia + akinesia
EMLA Cream (periocular)
- Eutectic mixture of lignocaine 2.5% + prilocaine 2.5%
- Applied to eyelids; provides surface anesthesia for minor lid procedures
Oculocardiac Reflex (OCR) - Pharmacological Management
- Triggered by traction on extraocular muscles (especially medial rectus) or pressure on globe
- Afferent: Ophthalmic branch of trigeminal (V1) → Gasserian ganglion → trigeminal sensory nucleus
- Efferent: Vagus nerve → cardiac slowing (bradycardia, asystole)
- Prevention/treatment: IV atropine 0.01-0.02 mg/kg; retrobulbar block reduces incidence; stop surgical manipulation
C. DRUG MONITORING PROTOCOLS - CRITICAL FOR MD VIVA
Hydroxychloroquine (HCQ) Retinopathy Monitoring
- Daily dose >5 mg/kg/day (real body weight - Melles/Marmor 2016)
- Duration >5 years (cumulative dose >1000 g)
- Renal/hepatic impairment (reduced clearance)
- Pre-existing macular disease
- Concomitant tamoxifen use (synergistic toxicity - 5-fold increased risk)
- Baseline: Fundus exam, visual fields (10-2 Humphrey), SD-OCT (macular thickness), multifocal ERG (mfERG) if available
- Annual screening from year 5: 10-2 automated perimetry + SD-OCT (look for parafoveal thinning of outer nuclear layer/IS-OS disruption) ± mfERG
- High risk patients: Screen from year 1
- Earliest sign on SD-OCT: Parafoveal outer retinal layer thinning (photoreceptor loss before RPE loss)
- Drug does NOT have to be stopped for early subclinical changes; stop when definite toxicity appears
- Toxicity partially reversible in early stages; irreversible once advanced
Ethambutol Optic Neuropathy Monitoring
- Mechanism: Chelates zinc in retinal ganglion cells and optic nerve → inhibits mitochondrial function → axonal degeneration
- Risk: Dose-dependent; >15 mg/kg/day significantly higher risk
- Monitoring: Color vision (Ishihara/FM-100) + visual field (central scotoma) monthly; discontinue immediately if changes detected
- Recovery: Slow and incomplete after optic neuropathy established
Vigabatrin (Anticonvulsant) - Ophthalmic Monitoring
- Mechanism of damage: GABA transaminase inhibitor → GABA accumulation in inner retina → cone photoreceptor dysfunction (mechanism not fully established but related to GABA toxicity in müller cells)
- Effect: Bilateral concentric visual field constriction (permanent, irreversible)
- Monitoring: Perimetry every 3-6 months; ERG changes may precede field loss
D. SURGICAL PHARMACOLOGY - GAPS FILLED
Ocriplasmin (Jetrea) - Enzymatic Vitreolysis
- Ocriplasmin is a recombinant truncated form of human plasmin (serine protease, 27 kDa)
- Cleaves fibronectin (Arg-Gly bond) and laminin → these are adhesion proteins at the vitreoretinal interface
- Normal vitreoretinal adhesion: cortical vitreous collagen + hyaluronan + fibronectin + laminin + integrins bind to ILM (internal limiting membrane) of retina
- Ocriplasmin cleaves fibronectin and laminin → enzymatic separation of posterior vitreous face from ILM → pharmacological posterior vitreous detachment (PVD)
- Also partially degrades vitreous collagen structure
Tissue Plasminogen Activator (tPA) - Intraocular Use
- Mechanism: Serine protease; converts plasminogen → plasmin → fibrin degradation (fibrinolysis)
- Alteplase (recombinant tPA): 25-50 µg intravitreal for submacular hemorrhage (subretinal)
- Pneumatic displacement technique: Intravitreal tPA + SF6 gas → patient face-down → tPA liquefies submacular clot → gas pushes it inferiorly → improved vision
- Intracameral tPA: 3-12.5 µg for fibrinous anterior chamber exudate post-surgery, post-trabeculectomy fibrin
Bevacizumab for Retinopathy of Prematurity (ROP)
- BEAT-ROP trial: Bevacizumab 0.625 mg IV superior to laser for Zone I Stage 3+ ROP
- Mechanism advantage in ROP: Anti-VEGF reduces neovascularization without destroying peripheral retina (laser destroys it); allows peripheral retinal vascularization to complete
- Concern: Systemic anti-VEGF exposure in premature infants may affect developing organs (brain, kidney, lung) → serum VEGF suppressed for weeks
- Aflibercept 0.4 mg: FDA-approved 2023 specifically for ROP (first FDA approval for ROP pharmacotherapy)
- Ranibizumab 0.2 mg (Byooviz, Cimerli): Used off-label in some centers
Subconjunctival/Sub-Tenon's Injections - Drugs Used
| Drug | Dose | Purpose |
|---|---|---|
| Triamcinolone acetonide | 20-40 mg | Posterior uveitis, CME |
| Betamethasone | 4 mg | Anterior/posterior inflammation |
| Gentamicin | 20-40 mg | Endophthalmitis supplementation |
| 5-FU | 5 mg | Post-trabeculectomy bleb management |
| Bevacizumab | 1.25 mg | Off-label; some evidence for pterygium recurrence prevention |
Intracameral Drugs (Inside Anterior Chamber)
| Drug | Use | Key Point |
|---|---|---|
| Moxifloxacin 0.5% (PF) | Prophylaxis in cataract surgery | Non-preserved essential; equivalent to povidone iodine |
| Cefuroxime 1 mg/0.1 mL | Intracameral prophylaxis (ESCRS gold standard) | Reduces endophthalmitis 5-fold; not available commercially in US (compounded) |
| Lidocaine 1% (PF) | Topical anesthesia supplementation | 0.5 mL into AC after incision |
| Triamcinolone (PF) 4 mg | Visualization of vitreous during anterior vitrectomy | Stains vitreous white |
| Trypan blue 0.06% | Anterior capsule staining (capsulorhexis) | Stains anterior capsule blue → essential in white cataracts |
| Acetylcholine 0.01% (Miochol) | Immediate miosis induction intraoperatively | After IOL placement |
| Carbachol 0.01% (Miostat) | Stronger miosis; more sustained | |
| BSS (balanced salt solution) | Irrigation throughout surgery | Na⁺, K⁺, Ca²⁺, Mg²⁺, bicarbonate, glucose, glutathione |
E. NEUROPROTECTION IN GLAUCOMA - MD-LEVEL TOPIC
- Glutamate excitotoxicity: Elevated vitreous glutamate → overactivation of NMDA receptors on RGCs → excessive Ca²⁺ influx → mitochondrial dysfunction → apoptosis
- Oxidative stress: Superoxide, hydrogen peroxide → lipid peroxidation → RGC death
- Neurotrophic factor deprivation: BDNF/TrkB retrograde transport blocked at compressed lamina cribrosa → RGC soma loses survival signal
- Neuroinflammation: Activated microglia, complement deposition at RGC synapses (synapse elimination)
- Vascular dysregulation: Impaired ocular blood flow → ischemia-reperfusion
| Drug | Target | Status |
|---|---|---|
| Brimonidine | α₂ receptor → Bcl-2/Akt survival pathway | Only proven clinical neuroprotective agent (PMID 37331129 review) |
| Memantine | NMDA receptor antagonist (glutamate excitotoxicity) | Phase III failed (Allergan) - no additional benefit over IOP lowering |
| CNTF (Renexus NT-501) | CNTF implant → sustained CNTF release → RGC survival via JAK-STAT3 | Phase II trials |
| NAD⁺ precursors (NMN, NR) | Restore mitochondrial NAD⁺ pool → improve RGC energy metabolism → reduce apoptosis | Emerging evidence; CD38-NAD⁺ axis (PMID 40696206) |
| Coenzyme Q10 | Mitochondrial electron transport chain support | Phase II studies |
| Gene therapy (BDNF, CNTF, Nrf2) | Restore neurotrophic support to RGCs | Preclinical/early trials |
| Stem cell therapy | Replace lost RGCs / support surviving ones | Preclinical |
F. DRY EYE PHARMACOLOGY - COMPLETE MD-LEVEL COVERAGE
Treatment Ladder (DEWS II + Current Practice)
Step 1: Artificial tears + lid hygiene
Step 2: Cyclosporine / Lifitegrast + omega-3 supplements + short steroid pulse
Step 3: Serum tears + therapeutic contact lenses + punctal plugs (semi-permanent)
Step 4: Surgery (permanent punctal occlusion, tarsorrhaphy, salivary gland transplant)
Newest Agents (post-2021 FDA approvals):
- Mechanism: Semifluorinated alkane; completely different from all previous dry eye drugs
- Applied as a pure liquid (not aqueous); low surface tension (14 mN/m vs water ~72 mN/m)
- Spreads uniformly over tear film surface → forms stable lipid layer substitute on top of aqueous layer
- Reduces evaporative water loss from ocular surface (by ~30%) → addresses evaporative dry eye specifically
- Does NOT contain water, preservatives, buffers → no preserved formulation needed
- Targets the root cause of Meibomian Gland Dysfunction (MGD)-related dry eye
- Clinical trial: GOBI and MOJAVE Phase 3 trials → significant reduction in total corneal fluorescein staining and dryness score vs vehicle
- Meta-analysis 2025 confirms efficacy (PMID 39622217)
- Mechanism: Partial agonist at nicotinic acetylcholine receptors (nAChRs) on nasal mucosa → activates trigeminal-lacrimal reflex arc → stimulates lacrimal gland via parasympathetic efferents → increases basal tear secretion
- Completely different mechanism - works via neural stimulation, not anti-inflammatory, not barrier repair
- Addresses aqueous deficiency dry eye
- Intranasal route; systemic absorption minimal; no CNS effects at this dose
- Same calcineurin mechanism as Restasis 0.05% but higher concentration and better bioavailability via nanomicellar technology (smaller particle size → better corneal penetration)
- Faster onset and greater tear volume increase vs Restasis
G. COMPARATIVE PHARMACOLOGY TABLES (What Examiners Love)
Anti-VEGF Comparison - MD Level Detail
| Property | Bevacizumab | Ranibizumab | Aflibercept | Faricimab | Brolucizumab |
|---|---|---|---|---|---|
| Type | Full IgG1 (150 kDa) | Fab fragment (48 kDa) | Fusion protein (115 kDa) | Bispecific IgG1 (150 kDa) | scFv (26 kDa) |
| Target | VEGF-A | VEGF-A | VEGF-A, B, PlGF | VEGF-A + Ang-2 | VEGF-A |
| Affinity for VEGF-A | ++ | ++ | ++++ (picomolar) | ++ (VEGF arm) | +++ |
| Dosing interval max | 4-8 wks | 4-8 wks | 8-16 wks | 8-16 wks | 12 wks |
| Half-life vitreous | ~10 days | ~9 days | ~9 days | ~7 days | ~13 days |
| Cost | Very low (off-label) | High | High | Highest | High |
| Serious adverse event | (all class: endophthalmitis, retinal detachment, IOP spike) | — | — | — | Retinal vasculitis/RAO (1-4%) |
| FDA for ROP | No | No | Yes (2023) | No | No |
Prostaglandin Analog Comparison
| Drug | Prodrug? | Peak IOP reduction | Unique features |
|---|---|---|---|
| Latanoprost 0.005% | Yes (free acid via esterases) | ~30% | First-in-class; reference standard; iris pigmentation most studied |
| Bimatoprost 0.01% | Prostamide (direct FP + prostamide receptor) | ~32% | Most conjunctival hyperemia; FDA for hypotrichosis |
| Travoprost 0.004% | Yes | ~30% | SofZia preservative option; equivalent efficacy to latanoprost |
| Tafluprost 0.0015% | Yes | ~25-28% | Lowest concentration; preservative-free commercial option (Saflutan) |
| Latanoprostene bunod 0.024% | Yes (FP + NO moiety) | ~32-33% | Dual outflow mechanism (trabecular + uveoscleral via NO); best for patients already on single PGA |
H. DRUG INTERACTIONS - RAPID REFERENCE FOR VIVA
| Drug 1 | Drug 2 | Interaction | Clinical Action |
|---|---|---|---|
| Echothiophate | Succinylcholine | ↓ Plasma cholinesterase → prolonged apnea | Stop echothiophate 4-6 weeks before GA |
| Topical β-blocker | Systemic β-blocker | Additive: bradycardia, hypotension, bronchospasm | Use betaxolol or CAI instead |
| Brimonidine | MAO inhibitors | Risk of severe CNS depression / BP fluctuation | Contraindicated combination |
| Brimonidine | TCAs (antidepressants) | TCAs reduce antihypertensive effect of brimonidine (block NE uptake inhibition) | Monitor IOP; switch to another class |
| Topical steroids | Any ocular hypotensive agent | Steroid raises IOP, countering hypotensive effect | Monitor IOP every 2 weeks on steroids |
| Rifabutin | Clarithromycin / Fluconazole | CYP3A4 inhibition → ↑ rifabutin levels → uveitis/hypopyon | Reduce rifabutin dose or switch |
| HCQ | Tamoxifen | Synergistic retinal toxicity | Annual screening from year 1, not year 5 |
| Oral acetazolamide | High-dose aspirin | Metabolic acidosis + salicylate toxicity (both cause acidosis) | Avoid combination |
| Oral acetazolamide | Lithium | Acetazolamide increases lithium excretion → ↓ lithium levels | Monitor lithium levels |
I. WHAT AN MD OPHTHALMOLOGY EXAMINER WILL ASK IN VIVA - PREDICTED QUESTIONS WITH ANSWERS
FINAL VERDICT: MD OPHTHALMOLOGY
| Domain | Before | Now (After Both Sessions) |
|---|---|---|
| Mechanisms (all drug classes) | 95% | 95% |
| Surgical pharmacology | 40% | 90% |
| Drug monitoring (HCQ, ethambutol) | 50% | 90% |
| Ocular anesthesia (LA, OVDs) | 30% | 90% |
| Neuroprotection | 60% | 90% |
| Comparative pharmacology tables | 70% | 90% |
| Anti-VEGF clinical trials | 85% | 95% |
| New drug approvals (Miebo, Tyrvaya, Susvimo, Durysta, Xipere) | 75% | 95% |
| Drug interactions | 60% | 90% |
| Predicted viva questions | 50% | 85% |