Hypersensitivity reaction pathology
Reading File
Finding Sources
Searching the Web
hypersensitivity reaction types pathology Gell Coombs classification mechanisms
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have comprehensive content. Let me compile the full answer from Robbins Pathologic Basis of Disease and Roitt's Essential Immunology.
Hypersensitivity Reactions - Pathology
Hypersensitivity reactions are immunologic reactions occurring with exaggerated or inappropriate intensity, directed against antigens that pose no real threat or against self-antigens, resulting in tissue damage (immunopathology). The underlying mechanisms are the same as those normally used to fight infection - they simply occur at the wrong time, place, or magnitude.
The Gell and Coombs classification (Types I-IV) remains the standard framework, with Type V (stimulatory) and innate hypersensitivity recognized as later additions.
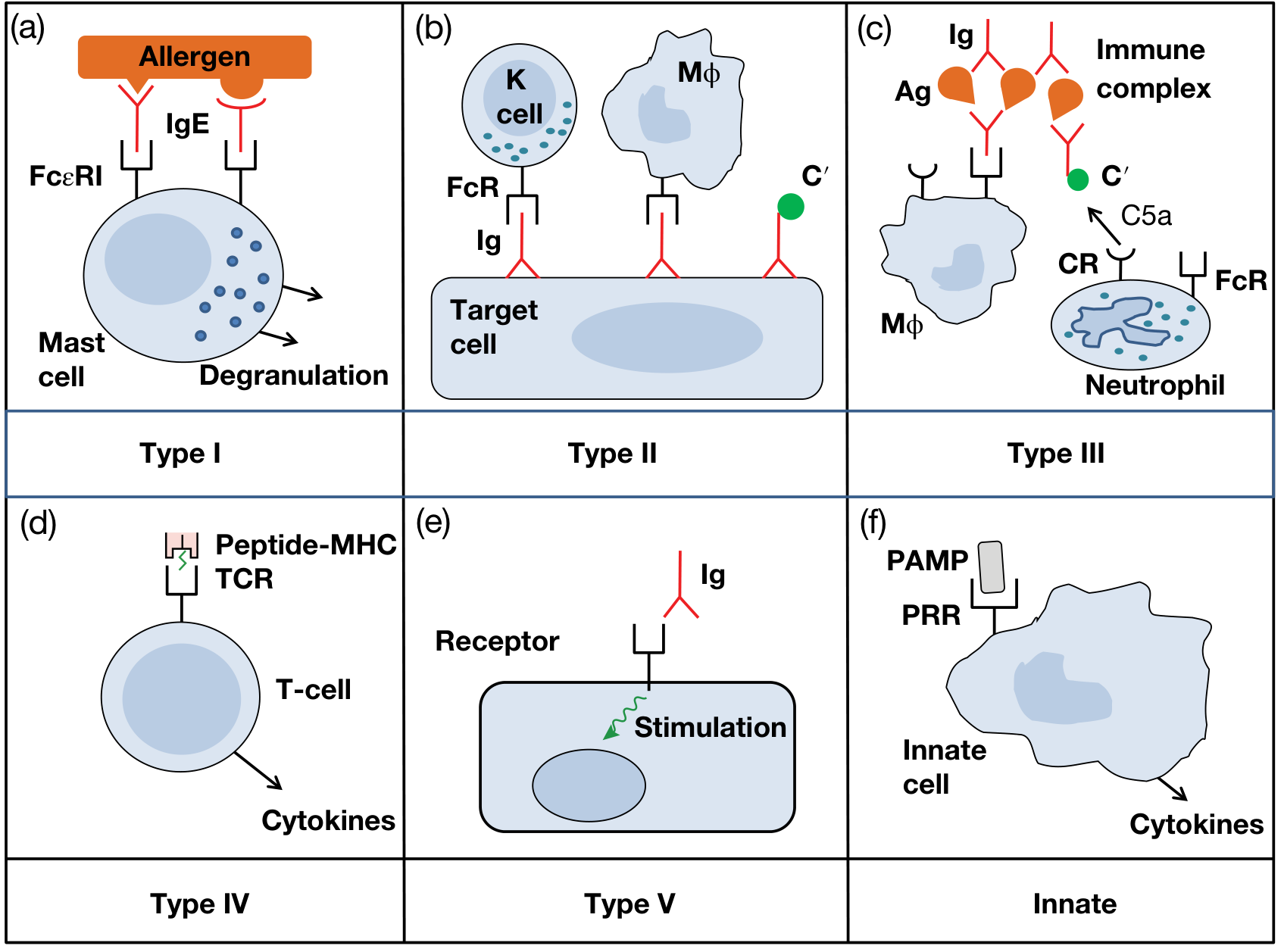
Fig. 14.1 from Roitt's Essential Immunology: Types I-IV (Gell-Coombs), Type V (stimulatory), and innate hypersensitivity
Summary Table (Robbins)
| Type | Mechanism | Histopathology | Prototypical Diseases |
|---|---|---|---|
| I - Immediate | IgE → mast cell degranulation → vasoactive mediators | Vascular dilation, edema, smooth muscle spasm, mucus production | Anaphylaxis, allergic asthma, urticaria, hay fever |
| II - Antibody-mediated | IgG/IgM binds cell/tissue antigen → complement activation, phagocytosis, ADCC | Phagocytosis/lysis of cells; inflammation; functional derangements | Autoimmune hemolytic anemia, Goodpasture syndrome |
| III - Immune complex | IgG-antigen complexes deposit in tissues → complement activation → leukocyte recruitment | Necrotizing vasculitis, fibrinoid necrosis | SLE, serum sickness, Arthus reaction, some glomerulonephritis |
| IV - Cell-mediated | Th1/Th17 cells release cytokines; CD8+ CTLs cause direct cytotoxicity | Perivascular infiltrates, granuloma formation, cell destruction | Contact dermatitis, tuberculosis, type 1 diabetes, MS |
Type I - Immediate (IgE-mediated) Hypersensitivity
Mechanism - Sensitization phase:
- First antigen exposure: APCs present allergen peptides to naive CD4+ T cells, which differentiate into Th2 cells under IL-4 signaling
- Th2 cells produce IL-4 and IL-13, which act on B cells to class-switch to IgE production
- IgE binds to FcεRI receptors on mast cells and basophils - the individual is now sensitized
Effector phase (re-exposure):
- Antigen cross-links IgE on mast cell surface → mast cell degranulation
- Primary (preformed) mediators: histamine, heparin, proteases (tryptase, chymase), eosinophil/neutrophil chemotactic factors
- Secondary (newly synthesized) mediators: prostaglandins, leukotrienes (LTC4, LTD4, LTE4), PAF, cytokines (IL-4, IL-5, IL-13, TNF)
Two-phase reaction:
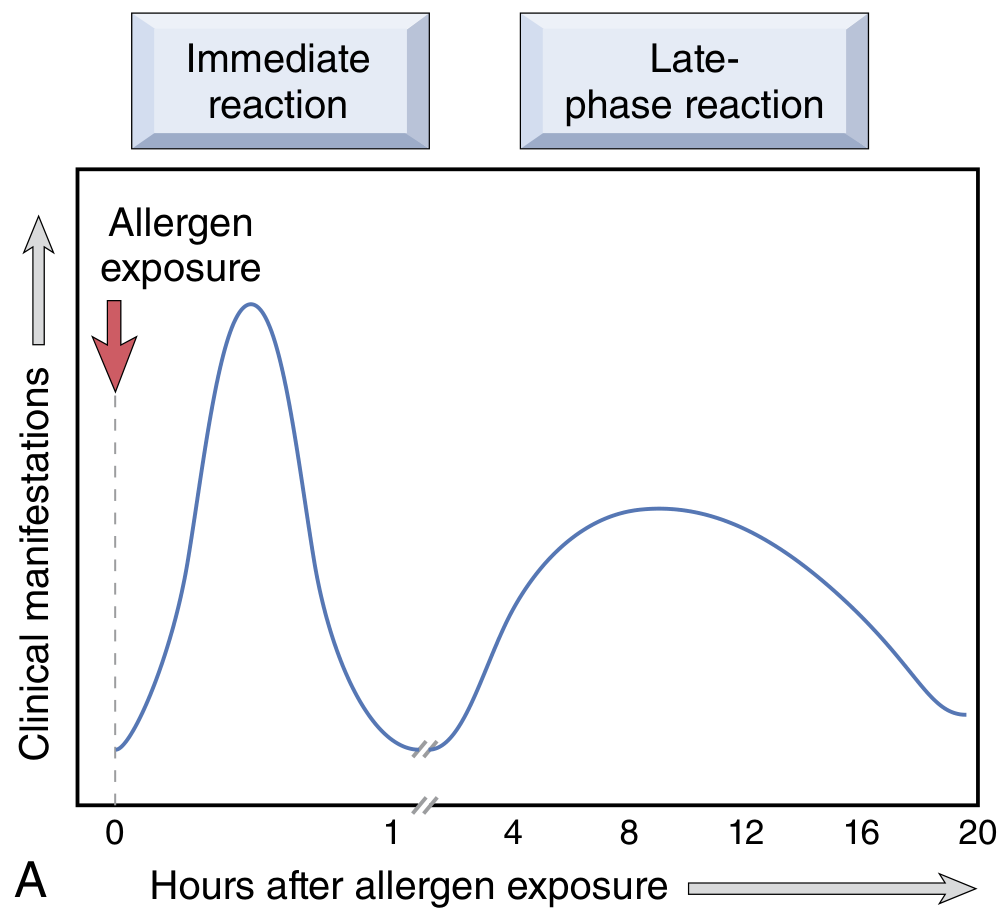
- Immediate reaction (within minutes): vasodilation, increased vascular permeability, smooth muscle spasm, glandular secretions - subsides within hours
- Late-phase reaction (2-24 hours later, without re-exposure): tissue infiltration with eosinophils, neutrophils, basophils, monocytes, CD4+ T cells; epithelial damage - persists for days
Clinical manifestations: systemic anaphylaxis (bee sting, IV penicillin), allergic rhinitis, asthma (atopic), urticaria, angioedema, food allergy
Histology: vascular congestion, edema, eosinophil-rich inflammatory infiltrate in the late phase
Type II - Antibody-Mediated (Cytotoxic) Hypersensitivity
Mechanism: IgG (rarely IgM) antibodies bind to antigens on cell surfaces or extracellular matrix
Three effector pathways:
- Complement-mediated lysis: IgG/IgM activates classical complement → MAC formation → cell lysis; C3b/C4b opsonization → phagocytosis
- ADCC (Antibody-Dependent Cell-mediated Cytotoxicity): NK cells, neutrophils, macrophages with FcR bind IgG-coated cells and kill them
- Functional alteration without tissue injury: antibodies against receptors alter normal cell signaling (e.g., anti-TSH receptor in Graves disease stimulates thyroid; anti-AChR in myasthenia gravis blocks neurotransmission)
Histology: phagocytosis/lysis of targeted cells; neutrophilic inflammation; in hemolytic disease, spherocytosis and extravascular hemolysis
Examples:
- Autoimmune hemolytic anemia, immune thrombocytopenic purpura
- Goodpasture syndrome (anti-GBM antibodies)
- Graves disease (stimulatory - sometimes classified Type V)
- Myasthenia gravis (blocking)
- Hemolytic transfusion reactions, erythroblastosis fetalis
- Rheumatic fever (anti-streptococcal Ab cross-reacts with cardiac myosin)
Type III - Immune Complex-Mediated Hypersensitivity
Mechanism: Soluble antigen-antibody complexes form in circulation or in situ, deposit in tissues, then activate complement and recruit leukocytes
Three sequential phases (Robbins):
- Immune complex formation: antigen excess favors small, soluble complexes that evade phagocytosis; slight antigen excess produces intermediate-sized complexes that are the most pathogenic
- Immune complex deposition: high-pressure filtration sites (glomeruli, joints, choroid plexus, skin) concentrate complexes - explains organ tropism
- Inflammation and injury: complement activation generates C3a/C5a (anaphylatoxins) → mast cell degranulation + neutrophil recruitment → release of lysosomal enzymes and ROS → tissue destruction
Hallmark histology: fibrinoid necrosis - smudgy eosinophilic destruction of vessel walls with neutrophilic infiltration; necrotizing vasculitis. Glomerular deposits appear as granular (lumpy-bumpy) pattern on immunofluorescence (distinguishes from Type II, which gives linear pattern).
Systemic (serum sickness) vs. Local (Arthus reaction):
- Serum sickness: 5-10 days after single large antigen exposure; fever, urticaria, joint pain, lymphadenopathy, proteinuria; serum C3 levels fall due to complement consumption
- Arthus reaction: localized skin necrosis after intradermal antigen injection in a pre-immunized individual; large complexes precipitate in vessel walls → fibrinoid necrosis + thrombosis
Examples: SLE, serum sickness, post-streptococcal GN, reactive arthritis, hypersensitivity pneumonitis, cryoglobulinemic vasculitis
Type IV - Cell-Mediated (Delayed-Type) Hypersensitivity
The only type NOT antibody-mediated. Divided into two subtypes:
IVa - Delayed-Type Hypersensitivity (DTH) via CD4+ T Cells
- Sensitization (1-2 weeks): antigen presented on MHC II → Th1/Th17 differentiation
- Elicitation (re-exposure, 48-72 hours later - hence "delayed"):
- Th1 cells release IFN-γ → macrophage activation → release of lysosomal enzymes, ROS, nitric oxide
- Th17 cells release IL-17 → neutrophil recruitment
- Persistent antigen → granuloma formation (hallmark of chronic DTH): epithelioid macrophages, multinucleated giant cells, surrounded by CD4+ T cells and fibroblasts
IVb - Direct Cytotoxicity via CD8+ CTLs
- Antigen presented on MHC I → CD8+ CTLs kill target cells directly via perforin/granzyme and Fas-FasL
Histology: perivascular mononuclear infiltrate (lymphocytes, macrophages); granulomas in chronic disease; no antibody/complement deposition
Examples:
- Contact dermatitis (poison ivy, nickel, latex) - prototype
- Tuberculosis - caseating granulomas
- Type 1 diabetes mellitus - CTL destruction of beta cells
- Multiple sclerosis - Th1/Th17 attack on myelin
- Transplant rejection (cellular component)
- Tuberculin skin test (PPD reaction at 48-72 hours)
Type V - Stimulatory Hypersensitivity (Addition to original classification)
Antibodies bind to cell-surface receptors and mimic ligand rather than destroy the cell. This is a functional disorder without cell injury - sometimes grouped under Type II but mechanistically distinct.
Examples:
- Graves disease: anti-TSH receptor IgG acts as TSH agonist → continuous thyroid stimulation → hyperthyroidism
- Contrasted with myasthenia gravis (Type II blocking) where the antibody inhibits receptor function
Key Distinctions to Remember
| Feature | Type I | Type II | Type III | Type IV |
|---|---|---|---|---|
| Mediator | IgE | IgG/IgM | IgG complexes | T cells |
| Onset | Minutes | Minutes-hours | Hours-days (10 days serum sickness) | 48-72 hours |
| Key cells | Mast cells, eosinophils | Complement, phagocytes, NK cells | Neutrophils (complement-recruited) | Macrophages, CTLs |
| Complement | No | Yes (classical) | Yes (classical) | No |
| IF pattern (kidney) | - | Linear | Granular | - |
| Passive transfer | Serum (IgE) | Serum (IgG) | Serum (complexes) | Cells only |
Note: Many diseases involve more than one type simultaneously. For example, SLE involves Type III (immune complexes) and potentially Type II (anti-RBC, anti-platelet antibodies). Seronegative RA has significant Type IV involvement alongside humoral mechanisms.
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 198-210 | Roitt's Essential Immunology, 13th ed., Ch. 14 | Harrison's Principles of Internal Medicine 22E
Define hypersensitivity from Robbins
Finding Sources
Reading File
Here is the definition of hypersensitivity directly from Robbins, Cotran & Kumar - Pathologic Basis of Disease:
Hypersensitivity - Definition (Robbins)
"Injurious immune reactions, called hypersensitivity, are responsible for the pathology associated with immunologic diseases. This term arose from the idea that individuals who have been previously exposed to an antigen will manifest detectable reactions to that antigen and are therefore said to be sensitized. Hypersensitivity implies an excessive or harmful reaction to an antigen."
Robbins goes on to outline four key general features:
-
Antigens can be exogenous or endogenous. Exogenous antigens include dust, pollen, food, drugs, microbes, and chemicals - reactions to these range from trivial (skin itching) to fatal (anaphylaxis). Reactions to self (autologous) antigens cause autoimmune diseases.
-
Hypersensitivity results from an imbalance between the effector mechanisms of immune responses and the control mechanisms that normally limit them. In many hypersensitivity diseases, the underlying cause is suspected to be a failure of normal regulation.
-
Genetic susceptibility plays a role. Development of hypersensitivity diseases (allergic and autoimmune) is often associated with inheritance of particular susceptibility genes - including HLA and non-HLA genes.
-
The mechanisms of tissue injury are the same as normal immune defense. The problem in hypersensitivity is that these reactions are poorly controlled, excessive, or misdirected - for example, against normally harmless environmental antigens or self antigens.
Source: Robbins, Cotran & Kumar - Pathologic Basis of Disease, Chapter 6: "Hypersensitivity: Immunologically Mediated Tissue Injury," p. 181
This is a shared conversation. Sign in to Orris to start your own chat.