Explain anemia with respect to pathology
Reading File
Finding Sources
Finding Sources
Searching PubMed
"anemia"[MeSH Terms] AND "pathology"[MeSH Terms]
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Finding Sources
Reading File
Loading Image
Finding Sources
Reading File
Loading Image
Loading Image
I now have comprehensive content from Robbins to construct a thorough, well-illustrated pathology explanation of anemia. Let me compile the full response:
Anemia - Pathology
Anemia is defined as a reduction in red cell mass (or hemoglobin concentration) below the normal range, impairing oxygen delivery to tissues. Normal hemoglobin thresholds are approximately <13.5 g/dL in men and <12 g/dL in women (lower limits vary by age).
1. Pathophysiologic Classification
All anemias arise from one or more of three fundamental mechanisms:
| Mechanism | Examples |
|---|---|
| Blood loss (hemorrhage) | Acute trauma, GI bleeding, menorrhagia |
| Increased red cell destruction (hemolysis) | Sickle cell, spherocytosis, autoimmune |
| Decreased red cell production (marrow failure) | Aplastic anemia, megaloblastic anemia, ACI |
- Robbins & Kumar Basic Pathology, p. 433
2. Morphologic Classification by MCV
A practical and widely used system categorizes anemia by red cell size:
| Type | MCV | Common Causes |
|---|---|---|
| Microcytic | <80 fL | Iron deficiency, thalassemia, anemia of chronic inflammation (ACI) |
| Normocytic | 80-100 fL | Aplastic anemia, acute blood loss, hemolytic anemia, early ACI |
| Macrocytic | >100 fL | Folate/B12 deficiency (megaloblastic), liver disease, high reticulocyte count |
3. Physiologic Compensation for Anemia
The body compensates via three mechanisms (Harrison's Principles of Internal Medicine 22E):
-
Increased cardiac output - occurs within minutes; oxygen delivery = cardiac output × hemoglobin, so the heart compensates rapidly when Hb falls. Patients with limited cardiac reserve become symptomatic at higher Hb levels.
-
Increased 2,3-DPG - occurs over hours to days. 2,3-DPG decreases hemoglobin's oxygen affinity, shifting the O2 dissociation curve rightward and promoting tissue O2 delivery despite lower circulating Hb.
-
Expanded plasma volume - occurs over weeks. Preserves cardiac output and blood pressure, but can precipitate high-output heart failure if severe.
4. Hemolytic Anemias
Hemolytic anemias share accelerated red cell destruction (life span reduced from the normal 120 days, often markedly so). The resulting tissue hypoxia stimulates EPO release, causing erythroid hyperplasia in the marrow and peripheral reticulocytosis - hallmarks of hemolysis. In severe cases, extramedullary hematopoiesis develops in the liver, spleen, and lymph nodes.
Extravascular vs. Intravascular Hemolysis
Extravascular hemolysis (most common) - red cells are destroyed by splenic macrophages due to reduced deformability or antibody coating. Features:
- Hyperbilirubinemia and jaundice (hemoglobin degraded in macrophages)
- Splenomegaly ("work hyperplasia" of splenic phagocytes)
- Pigment (bilirubin) gallstones if long-standing
Intravascular hemolysis - red cells burst within the circulation (e.g., mechanical trauma from a defective valve, complement fixation, clostridial toxins). Features:
- Hemoglobinemia, hemoglobinuria, hemosiderinuria
- Iron deficiency with chronic hemolysis (urinary iron loss)
- Decreased serum haptoglobin (in both types)
Robbins & Kumar Basic Pathology, pp. 384-385
4a. Sickle Cell Anemia
A point mutation in the β-globin gene substitutes valine for glutamate at position 6, producing HbS. Deoxygenated HbS polymerizes into long fibers that distort red cells into a sickle shape.
Peripheral blood smear - sickle cell anemia:
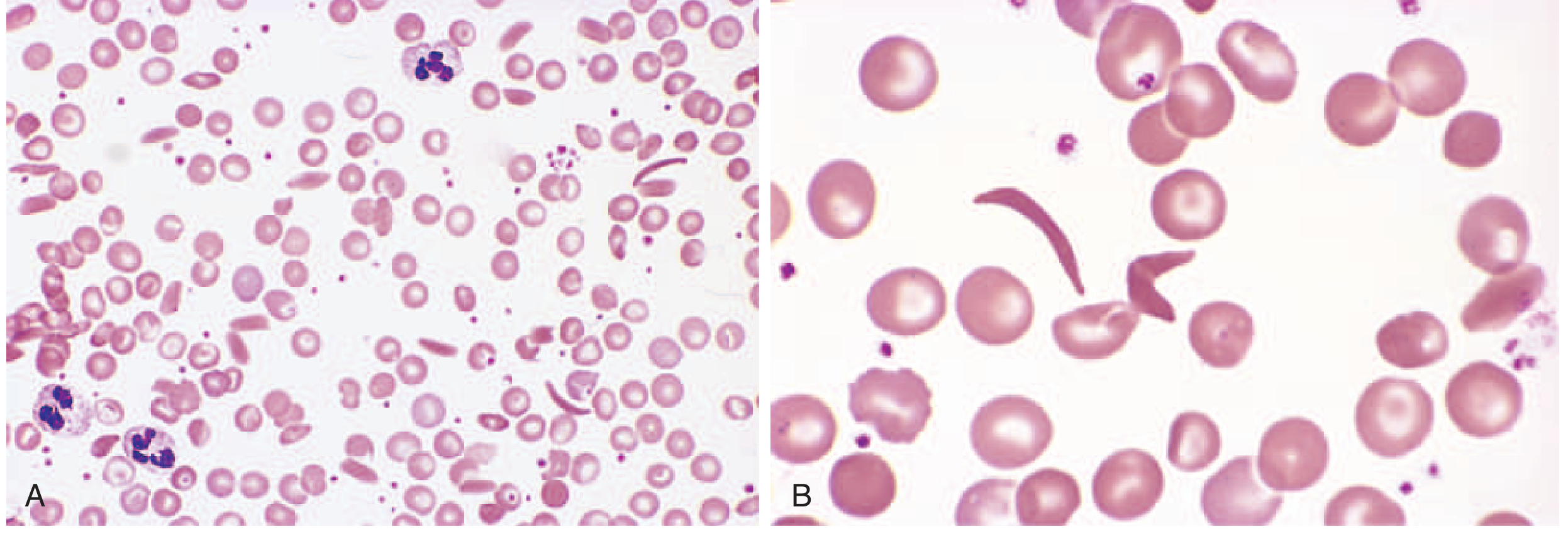
Panel A (low power): mixture of sickled and normal RBCs with nucleated red cells. Panel B (high power): classic elongated crescent-shaped sickled cells.
Key pathogenetic factors:
- HbF (fetal hemoglobin) inhibits HbS polymerization; symptoms don't appear until ~5-6 months of age
- Heterozygotes (sickle trait, ~40% HbS) are largely protected in vivo
- Repeated sickling episodes cause cumulative membrane damage, creating irreversibly sickled cells
Consequences include moderate-to-severe hemolytic anemia, vaso-occlusive crises (painful infarcts in bone, spleen, kidneys, brain), autosplenectomy, and susceptibility to encapsulated bacteria (Streptococcus pneumoniae, Haemophilus influenzae).
- Robbins & Kumar Basic Pathology, pp. 386-390
4b. Hereditary Spherocytosis
An autosomal dominant disorder caused by mutations that destabilize the red cell membrane skeleton (spectrin, ankyrin, band 3). The red cell loses membrane surface area relative to volume, forming a spherocyte - unable to navigate splenic sinusoids and phagocytosed. Presents with anemia, splenomegaly, and cholelithiasis.
4c. G6PD Deficiency
X-linked; deficiency of glucose-6-phosphate dehydrogenase renders red cells vulnerable to oxidant damage. Episodes of intravascular hemolysis are triggered by oxidant stressors: infections, drugs (primaquine, dapsone), and fava beans. Heinz bodies (denatured Hb precipitates) are seen.
4d. Immunohemolytic Anemia (Autoimmune Hemolytic Anemia)
Antibodies against normal or drug-modified red cell constituents drive premature destruction. Classified by antibody characteristics:
- Warm antibody type (IgG, active at 37°C) - 80% of cases: Idiopathic or secondary to SLE, lymphoid neoplasms, or drugs. IgG-coated cells bind Fc receptors on splenic macrophages → partial phagocytosis → spherocyte formation → extravascular hemolysis. Moderate splenomegaly.
- Cold agglutinin type (IgM, active <37°C): Seen in Mycoplasma infection, infectious mononucleosis, and lymphoid tumors. IgM activates complement; agglutination in cool peripheral vessels.
- Cold hemolysis type (IgG, active <37°C): Rare, mainly in children post-viral infections.
Diagnosis: Direct Coombs (antiglobulin) test - patient's red cells mixed with anti-IgG/anti-complement antibodies, causing agglutination if red cells are coated.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 607-611
5. Iron Deficiency Anemia
The most common nutritional deficiency worldwide - affects ~10% in high-income countries and 25-50% in low-income countries.
Iron Metabolism Regulation (Hepcidin-Ferroportin Axis):
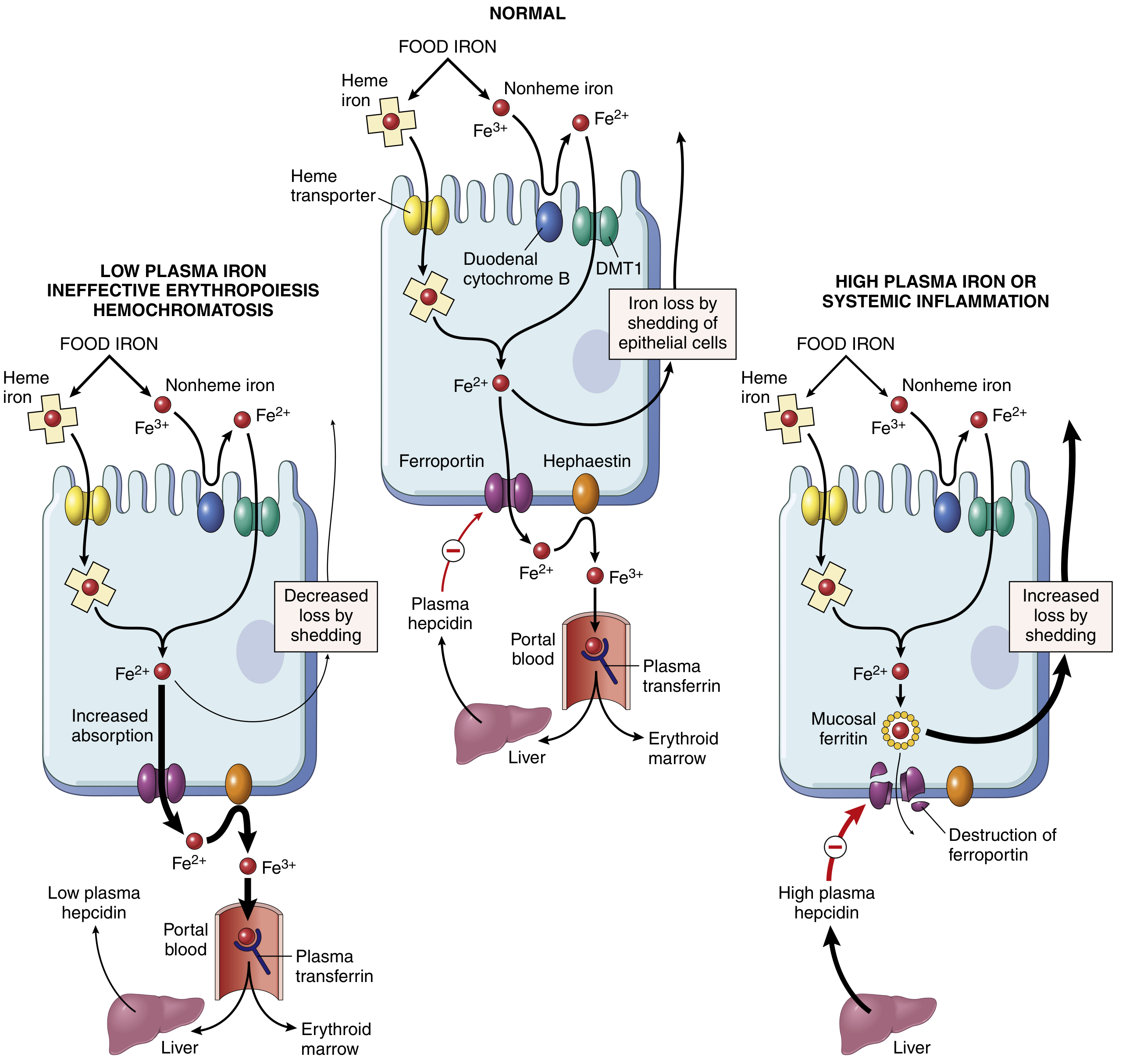
Hepcidin, secreted by the liver, degrades ferroportin and blocks iron export from intestinal enterocytes and macrophages. In iron deficiency or ineffective erythropoiesis, hepcidin falls and absorption increases (left panel). In high iron states or systemic inflammation, hepcidin rises and iron absorption is blocked (right panel).
Causes of iron deficiency:
- Increased demand: Pregnancy, infancy, growth
- Inadequate intake: Poor diet, malnutrition
- Impaired absorption: Celiac disease, gastrectomy, achlorhydria
- Chronic blood loss: GI ulcers/carcinoma, menorrhagia (most common cause in premenopausal women in high-income countries)
Iron Deficiency Peripheral Blood Smear:
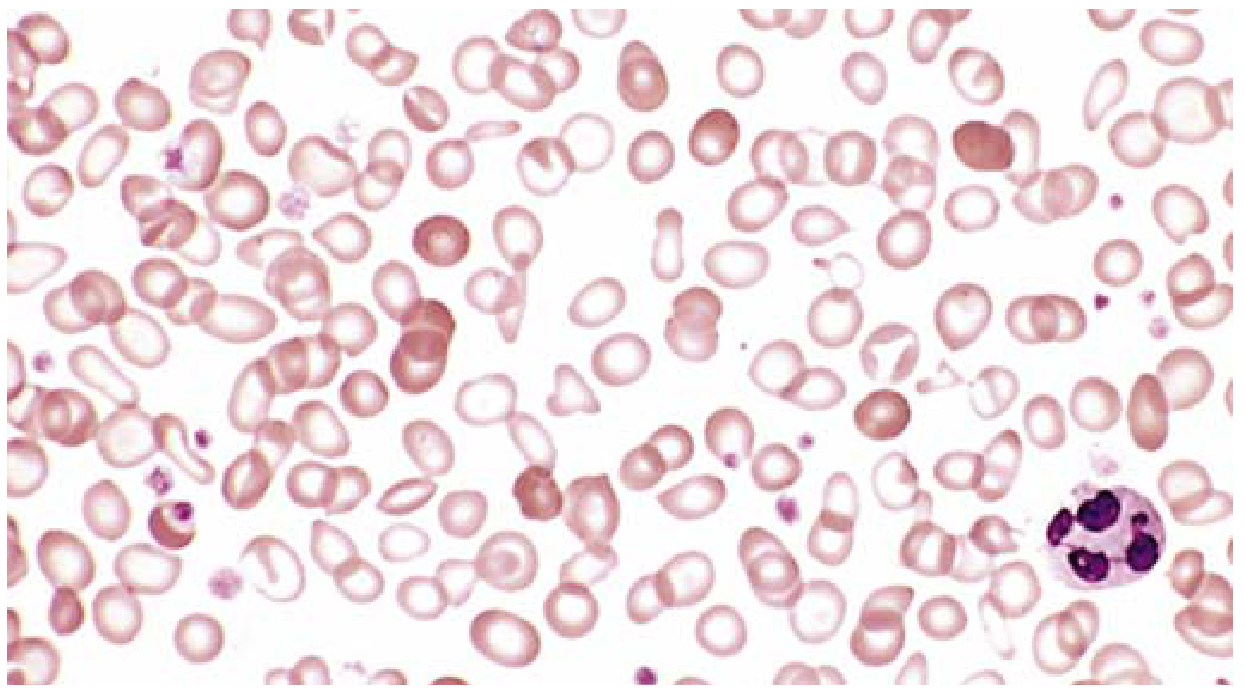
Characteristic features: microcytes, hypochromia (expanded area of central pallor >1/3 of cell diameter), pencil cells (elongated forms), anisocytosis and poikilocytosis. Scattered normally colored cells are from a prior transfusion.
Stages of iron deficiency:
- Depletion of iron stores (low ferritin, normal Hb)
- Iron-deficient erythropoiesis (low serum iron, elevated TIBC, transferrin saturation <16%)
- Iron deficiency anemia - overt microcytic, hypochromic anemia (MCV <80 fL)
Lab findings: Low serum iron, low ferritin, elevated TIBC, elevated red cell distribution width (RDW). Normal serum ferritin is about 12-300 ng/mL in adults; in iron deficiency it falls below 12 ng/mL.
- Robbins & Kumar Basic Pathology, pp. 393-395
6. Anemia of Chronic Inflammation (ACI)
Also called "anemia of chronic disease," ACI is seen in:
- Chronic infections: osteomyelitis, bacterial endocarditis, lung abscess
- Chronic immune disorders: rheumatoid arthritis, Crohn disease
- Malignancies: Hodgkin lymphoma, carcinomas of the lung and breast
Pathogenesis centers on elevated plasma hepcidin, driven by pro-inflammatory cytokines (especially IL-6). High hepcidin degrades ferroportin on bone marrow macrophages, trapping iron and blocking its transfer to erythroid precursors. Additionally, chronic inflammation suppresses renal EPO synthesis, further dampening erythropoiesis.
Key distinguishing features from iron deficiency:
| Feature | Iron Deficiency Anemia | Anemia of Chronic Inflammation |
|---|---|---|
| Serum iron | Low | Low |
| Serum ferritin | Low | Normal to high |
| TIBC / transferrin | High | Low |
| Bone marrow iron stores | Absent | Present/Increased |
| Red cell morphology | Microcytic, hypochromic | Normocytic/normochromic (or mild microcytic) |
Treatment requires addressing the underlying condition; erythropoietin + iron may be adjuncts.
- Robbins & Kumar Basic Pathology, p. 394; The Washington Manual of Medical Therapeutics
7. Megaloblastic Anemias
Both folate and vitamin B12 are required as cofactors for thymidine synthesis and DNA replication. Deficiency causes a block in cell division with normal cytoplasmic growth - termed nuclear-cytoplasmic asynchrony.
Morphology (common to all megaloblastic anemias):
- Bone marrow: markedly hypercellular; megaloblasts with fine, "open" chromatin (nuclear immaturity despite cytoplasmic maturation); giant metamyelocytes
- Peripheral blood:
- Macro-ovalocytes (large, oval RBCs; MCV often >110 fL)
- Hypersegmented neutrophils (5+ lobes) - earliest finding, appears before anemia
- Large, misshapen platelets
- Anisocytosis and poikilocytosis
Folate Deficiency
- Causes: poor diet (alcoholism, elderly), increased demand (pregnancy, hemolytic anemia), drugs (methotrexate, phenytoin), malabsorption (celiac disease)
- Stores last only 3-4 months
- No neurologic manifestations
Vitamin B12 Deficiency (Pernicious Anemia)
-
Most common cause: Pernicious anemia - autoimmune destruction of gastric parietal cells → loss of intrinsic factor (IF) → failure of ileal B12 absorption
-
Other causes: strict veganism, gastrectomy, ileal disease, bacterial overgrowth
-
Stores last 3-5 years before deficiency appears
-
Neurologic manifestations (subacute combined degeneration of spinal cord): demyelination of posterior and lateral columns → paresthesias, ataxia, weakness (folate deficiency does NOT cause this)
-
Robbins & Kumar Basic Pathology, pp. 395-397; Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 608-614
8. Aplastic Anemia
A disorder of multipotent myeloid stem cell suppression resulting in pancytopenia and a marrow largely devoid of hematopoietic elements (replaced by fat cells).
Pathogenesis - two major models:
- Immune-mediated (primary mechanism): Activated Th1 lymphocytes attack hematopoietic stem cells (possibly after antigen alteration by drugs/infections), producing cytokines (IFN-γ, TNF) that suppress HSCs. Immunosuppressive therapy restores hematopoiesis in 60-70% of patients.
- Intrinsic stem cell defect: 5-10% have inherited mutations in telomerase genes; ~50% more have abnormally short telomeres, leading to premature HSC senescence.
Clinical features:
- Insidious development of pallor, fatigue, dyspnea (anemia)
- Petechiae and ecchymoses (thrombocytopenia)
- Serious infections (neutropenia)
- No splenomegaly (distinguishes it from myelophthisic anemia and leukemia)
Treatment: HSC transplantation (curative in <40 years), or immunosuppression (anti-thymocyte globulin, cyclosporine).
- Robbins & Kumar Basic Pathology, pp. 400-401
9. Myelophthisic Anemia
Caused by extensive infiltration of the marrow by tumors or other lesions (most commonly metastatic breast, lung, or prostate cancer). The marrow space is physically replaced, impairing all cell lines. Peripheral blood shows:
- Leukoerythroblastosis (immature WBC and nucleated RBC precursors in blood)
- Teardrop cells (dacrocytes)
- Mild leukocytosis; thrombocytopenia
10. Summary Diagram of Classification
ANEMIA
|
┌────────────────────┼────────────────────┐
│ │ │
Blood Loss Hemolysis Decreased Production
│ │ │
Acute/ Intravascular/ Iron deficiency
Chronic Extravascular Megaloblastic
│ Aplastic anemia
┌────────┴───────┐ Myelophthisic
Intrinsic Extrinsic Anemia of
defects defects inflammation
(HS, SCA, G6PD, (Immune, TTP,
Thalassemia) Malaria)
Clinical Manifestations
The presentation depends on both the rate of onset and the cause:
- Acute anemia (e.g., large hemorrhage): shortness of breath, shock, organ failure
- Chronic anemia: insidious pallor, fatigue, lassitude, tachycardia, flow murmur
- Extravascular hemolysis: jaundice, splenomegaly, pigment gallstones
- Ineffective erythropoiesis (e.g., thalassemia major): iron overload (hemochromatosis) → cardiac and endocrine failure; bone deformities from marrow hyperplasia
Sources: Robbins & Kumar Basic Pathology (9e), Robbins Cotran & Kumar Pathologic Basis of Disease (10e), Harrison's Principles of Internal Medicine 22E (2025), The Washington Manual of Medical Therapeutics.
This is a shared conversation. Sign in to Orris to start your own chat.