Wilson disease is most prevalent in which age, what raises the suspicion of wilson
Reading File
Finding Sources
Searching PubMed
"Wilson disease" diagnosis clinical features
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information from multiple textbook sources. Here is the complete answer:
Wilson Disease
Age of Prevalence
Wilson disease (WD) is a disease of young people. Symptoms typically appear between 6 and 40 years of age, though onset as late as the fifth or sixth decade has been reported in rare cases. The most common presentation is in the second decade of life (adolescence/young adulthood). An acute presentation - often as acute liver failure - typically occurs in the second decade and accounts for up to 25% of cases.
- Robbins & Kumar Basic Pathology, p. 615
- Bradley & Daroff's Neurology in Clinical Practice, p. 3800
What Raises Suspicion of Wilson Disease?
1. Clinical Presentation Pattern
About one-third of patients present in each of three ways:
- Hepatic disease (acute hepatitis, chronic hepatitis, or cirrhosis)
- Neurological features (extrapyramidal: tremor, chorea, dystonia, dysarthria, dysphagia, ataxia, gait disturbance)
- Psychiatric symptoms (personality/mood changes, depression ~30%, bipolar features ~20%, psychosis)
Any young patient (especially <40 years) with unexplained liver disease plus neuropsychiatric features should raise immediate suspicion.
2. The Kayser-Fleischer (KF) Ring
The hallmark clinical sign. A yellow-brown/green-brown copper deposit in the Descemet membrane at the limbus of the cornea.
- Present in 98% of patients with neurological disease
- Present in ~80% of all cases of WD
- Best seen on slit-lamp examination
- Nearly all patients with neurological involvement have KF rings
3. Specific Clue in Acute Liver Failure (ALF)
When a young patient presents with ALF, Wilson disease is suspected when:
- Serum alkaline phosphatase : total bilirubin ratio < 4 (low ALP is unusual for most liver diseases)
- AST : ALT ratio > 2.2
- Unconjugated hyperbilirubinemia from hemolysis (Coombs-negative hemolytic anemia)
- Ascites as an early feature (suggesting underlying cirrhosis)
4. Laboratory Red Flags
| Test | Finding in Wilson Disease |
|---|---|
| Serum ceruloplasmin | Low (hallmark, though not the most sensitive) |
| 24-hour urinary copper | Elevated (most specific test) |
| Hepatic copper content | >250 µg/g dry weight (most sensitive; diagnostic at this level) |
| Serum copper | Unreliable - may be low, normal, or high depending on disease stage |
| Serum ALP | Paradoxically low in acute Wilson ALF |
5. Organ Systems Affected - Red Flags by System
| System | Suspicious Finding |
|---|---|
| Liver | Unexplained hepatitis, steatosis, cirrhosis in a young person |
| Neuro | Tremor, dystonia, dysarthria, ataxia in young age |
| Eye | Kayser-Fleischer rings on slit-lamp |
| Psychiatric | Personality change, depression, psychosis in a young person with hepatic disease |
| Blood | Coombs-negative hemolytic anemia (copper causes RBC hemolysis) |
| Kidney/Bone | Renal tubular acidosis, osteoporosis, fractures in young patients |
Pathophysiology Summary
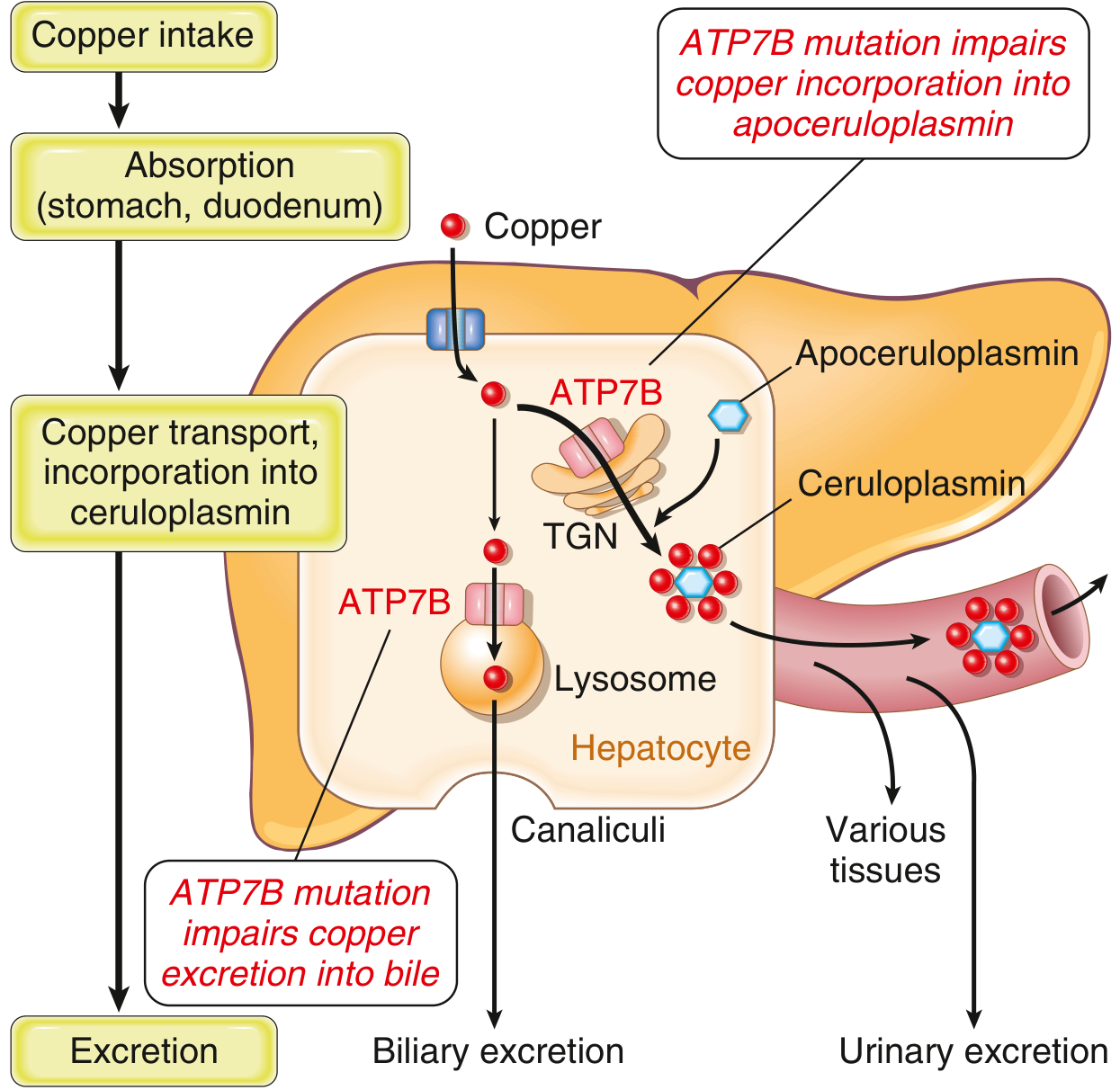
Fig. 14.22 from Robbins & Kumar: ATP7B mutation impairs both biliary copper excretion and ceruloplasmin formation, causing copper accumulation in hepatocytes and downstream toxicity to brain, corneas, kidneys, and bones.
WD is autosomal recessive, caused by loss-of-function mutations in ATP7B (chromosome 13), a copper-transporting ATPase. The result is:
- Copper cannot be excreted into bile (primary copper excretion route)
- Copper cannot be incorporated into apoceruloplasmin → low serum ceruloplasmin
- Hepatic copper accumulates → hepatocyte injury → copper spills into blood
- Free copper deposits in brain (basal ganglia), corneas, kidneys, joints, parathyroid glands
Sources: Robbins & Kumar Basic Pathology, pp. 614-616 | Bradley & Daroff's Neurology in Clinical Practice, p. 3800 | Sleisenger & Fordtran's GI and Liver Disease, p. 1838


IMG_2090.jpg
IMG_2091.jpg
Analyze this report
analyse_media
analyse_media
Here is a structured analysis of the two CT films:
CT Scan Analysis
Technical Details
| Parameter | Finding |
|---|---|
| Modality | CT (Computed Tomography) |
| Facility | EKO Diagnostics |
| Scan Date | 05 July 2020 |
| Patient | Nilanjana Ghosh (female) |
| Coverage | Full abdomen and pelvis (lower chest to proximal thighs) |
| Contrast | IV contrast used (vascular and parenchymal enhancement visible); oral contrast also administered (bright bowel loops in pelvis) |
| Views | Axial slices (sequential, cranio-caudal) + coronal reconstructions (bottom-left of first film) |
Film 1 (IMG_2090) - Overview: Chest Base + Full Abdomen + Pelvis
Left panel - Lower abdomen and pelvis (axial + coronal):
- Lumbar vertebrae and sacrum intact, no gross destructive lesions
- Bowel loops with normal gas distribution - no signs of obstruction
- Urinary bladder visible anteriorly in the pelvis
- Uterus/adnexa visible in mid-pelvis (female patient)
- Rectum seen posteriorly
- Psoas and abdominal wall musculature appear symmetric
- Coronal reconstructions (bottom rows) show the retroperitoneum and para-aortic region
Right panel - Upper abdomen (chest base to mid-abdomen):
- Liver: Well-visualized in the right upper quadrant with homogeneous enhancement; portal and hepatic venous branches visible
- Spleen: Normal crescentic shape, homogeneous enhancement, left upper quadrant
- Pancreas: Visible anterior to the splenic vein in the retroperitoneum
- Kidneys: Both kidneys show symmetric cortical/medullary enhancement (nephrographic phase); no obvious stones or masses at this resolution
- Stomach: Partially distended, normal
- Heart and lung bases: Visible in the upper slices; no obvious large pleural effusions or consolidation
Film 2 (IMG_2091) - Lower Abdomen and Pelvis Detail
This film shows sequential axial slices from the mid-abdomen down to the bony pelvis, providing finer detail of:
- The aortic bifurcation and common iliac vessels
- Bowel loops in the lower abdomen - normal distribution without distension or obstruction
- Pelvic inlet and true pelvis including the bladder, uterus, and rectum
- Bony pelvis: Iliac wings, sacrum, and femoral heads appear intact
- No gross free fluid (ascites) visible
- No large solid masses or cystic lesions apparent
Overall Impression (Visual Analysis)
| System | Observation |
|---|---|
| Liver | Appears homogeneous, no obvious focal lesion at this resolution |
| Spleen | Normal in size and attenuation |
| Kidneys | Symmetric enhancement, no obvious obstruction or mass |
| Bowel | Normal gas pattern, no obstruction |
| Pelvis | Bladder, uterus, and rectum appear grossly normal |
| Bones | No obvious lytic or sclerotic lesions |
| Vasculature | Aorta and IVC appear patent |
| Free fluid | No obvious ascites |
Important Limitations
These images are photographs of physical CT films on a light box, not high-resolution DICOM files. As a result:
- Fine details such as small renal stones, subtle liver lesions (<1 cm), small lymph nodes, or early peritoneal deposits cannot be reliably assessed
- Bone and lung windows are not separately available
- No definitive diagnosis can be made from this format
This analysis is for educational/orientation purposes only. A formal radiological report requires review of original DICOM images by a licensed radiologist with full clinical context.
This is a shared conversation. Sign in to Orris to start your own chat.