Biotransformation pharmacology
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive content from multiple authoritative textbooks. Let me compile the full answer.
Biotransformation in Pharmacology
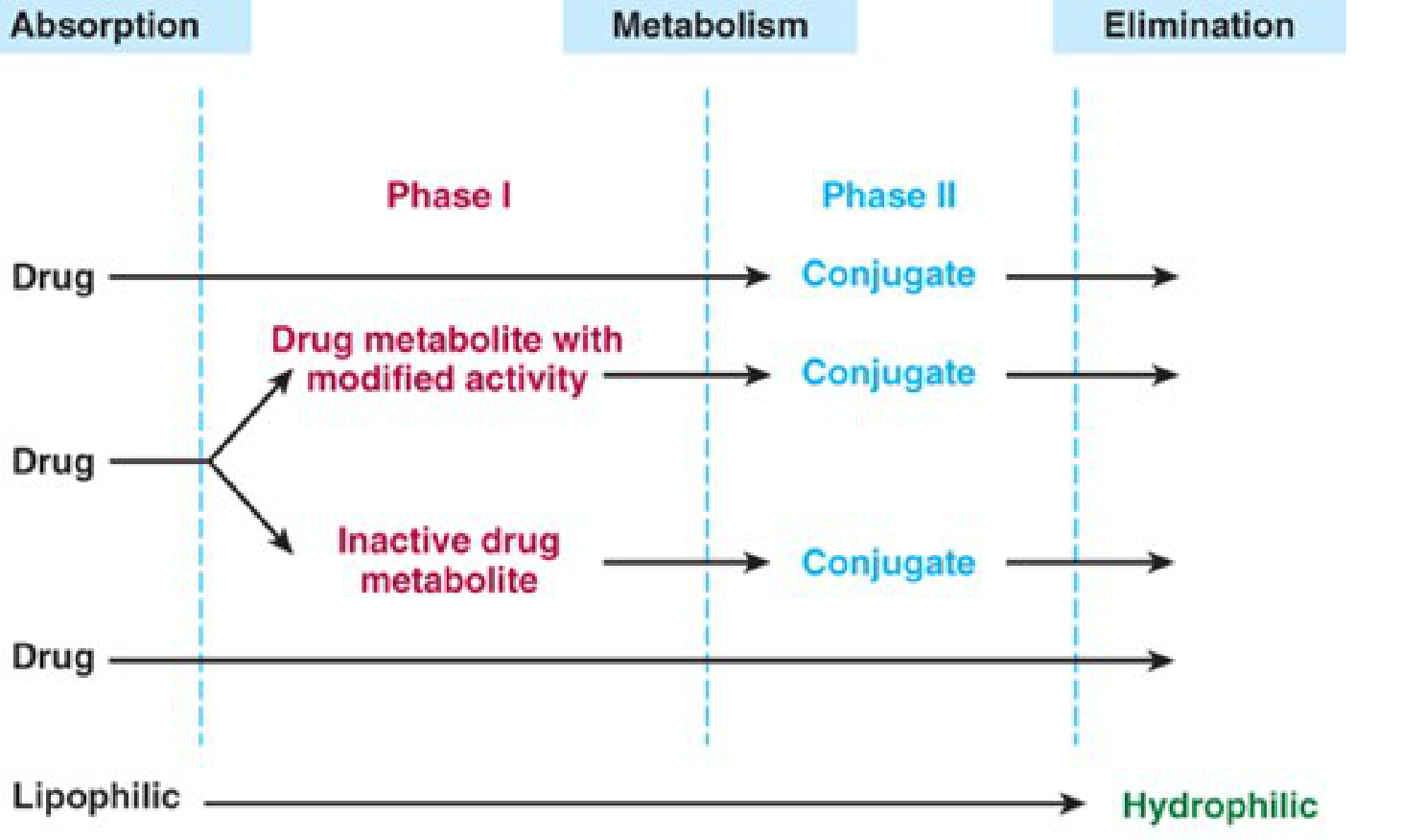
Biotransformation is the sum of chemical processes by which the body modifies a drug molecule - primarily converting lipophilic compounds into more polar, water-soluble forms that can be excreted in urine or bile.
1. Sites of Biotransformation
The liver is the principal organ of drug metabolism. Other tissues with significant capacity include:
- Gastrointestinal tract (esophagus, stomach, small intestine, colon)
- Lungs, skin, kidneys, brain
- Plasma (for esters, which are hydrolyzed there)
After oral administration, drugs absorbed from the small intestine travel via the portal vein to the liver before reaching systemic circulation - this is the first-pass effect (pre-systemic metabolism). Examples of high first-pass drugs: isoproterenol, meperidine, morphine, nitroglycerin. (Katzung's Basic & Clinical Pharmacology, 16th Ed.)
In the GI tract, CYP enzyme expression is highest in the duodenum and jejunum, declining progressively distally. Intestinal CYP3A4 is notably responsible for the grapefruit juice interaction - grapefruit furanocoumarins "knock out" intestinal (but not hepatic) CYP3A4, dramatically increasing bioavailability of substrates like verapamil, cyclosporine, and midazolam. (Yamada's Textbook of Gastroenterology, 7th Ed.)
2. Phase I Reactions
Phase I reactions introduce or unmask a polar functional group (-OH, -NH₂, -SH) through oxidation, reduction, or hydrolysis, producing more polar metabolites.
- Metabolites may be inactive, have modified activity, or even be more active than the parent drug
- If sufficiently polar, phase I metabolites may be excreted directly
- Otherwise, they proceed to Phase II
The Cytochrome P450 (CYP) System
The dominant Phase I enzymes are the CYPs - membrane-bound hemoproteins with peak light absorbance at 450 nm, located in the smooth endoplasmic reticulum of hepatocytes. 57 CYP genes exist in humans, but only ~10 are clinically relevant to drug metabolism.
Catalytic cycle: The substrate binds to the ferric (Fe³⁺) P450, which accepts an electron from NADPH-cytochrome P450 reductase, then binds O₂. A second electron is introduced, one oxygen atom is reduced to water, and the other is inserted into the substrate:
P450 + RH + O₂ + NADPH → P450 + R-OH + H₂O + NADP⁺
The potent oxidizing properties of this "activated oxygen" permit oxidation of a vast range of substrates.
Key CYP Isoforms and Their Clinical Significance
| CYP | Notable Substrates | Inducers | Inhibitors |
|---|---|---|---|
| 1A2 | Caffeine, theophylline, clozapine, warfarin, tacrine | Chargrilled/smoked meat, cigarette smoke, omeprazole, rifampin | Ciprofloxacin, fluvoxamine, cimetidine |
| 2A6 | Nicotine (→cotinine), acetaminophen, halothane | Rifampin, phenobarbital | Ketoconazole, isoniazid |
| 2B6 | Bupropion, methadone, efavirenz, ketamine, propofol | Carbamazepine, phenobarbital, rifampin, St. John's wort | Amiodarone, fluoxetine |
| 2C8 | Paclitaxel, rosiglitazone, repaglinide | Rifampin, barbiturates | Gemfibrozil, trimethoprim |
| 2C9 | Warfarin, phenytoin, NSAIDs (ibuprofen, diclofenac), losartan, sulfonylureas | Rifampin, carbamazepine | Fluconazole, amiodarone |
| 2C19 | Omeprazole, diazepam, clopidogrel (prodrug activation) | Rifampin | Omeprazole, fluvoxamine |
| 2D6 | Codeine (→morphine), TCAs, β-blockers, SSRIs, antipsychotics | Rifampin | Fluoxetine, quinidine, paroxetine |
| 3A4/5 | ~30% of all drugs: statins, cyclosporine, tacrolimus, midazolam, nifedipine, erythromycin | Rifampin, carbamazepine, St. John's wort | Ketoconazole, clarithromycin, grapefruit |
(Katzung's Basic & Clinical Pharmacology, 16th Ed.; Yamada's Textbook of Gastroenterology, 7th Ed.)
CYP3A4 is the most abundant single hepatic and intestinal CYP, involved in ~30% of clinically used drugs. Intestinal CYP3A4 expression is independent of hepatic expression, regulated by separate mechanisms.
Other Phase I Enzymes
- Flavin monooxygenases (FMOs): Oxidize nitrogen- and sulfur-containing drugs; complement CYP activity
- Monoamine oxidase (MAO): Degrades biogenic amines
- Epoxide hydrolases: Hydrolyze reactive epoxide intermediates generated by P450s
- Alcohol and aldehyde dehydrogenases
3. Phase II Reactions (Conjugation)
Phase II reactions conjugate the parent drug or its Phase I metabolite with an endogenous polar substrate, forming highly water-soluble conjugates for renal or biliary elimination.
Phase II reactions are generally faster than Phase I reactions. (Katzung's Basic & Clinical Pharmacology, 16th Ed.)
| Reaction | Enzyme | Co-substrate | Examples |
|---|---|---|---|
| Glucuronidation | UDP-glucuronosyltransferases (UGTs) | UDP-glucuronic acid | Morphine, bilirubin, paracetamol, NSAIDs |
| Sulfation | Sulfotransferases (SULTs) | 3'-phosphoadenosine-5'-phosphosulfate (PAPS) | Acetaminophen, minoxidil (activation!) |
| Acetylation | N-acetyltransferases (NAT1, NAT2) | Acetyl-CoA | Isoniazid, procainamide, hydralazine |
| Methylation | Methyltransferases (COMT, TPMT) | S-adenosylmethionine (SAM) | Dopamine, norepinephrine, 6-mercaptopurine |
| Glutathione conjugation | Glutathione-S-transferases (GSTs) | Glutathione (GSH) | Acetaminophen toxic metabolite (NAPQI) |
| Amino acid conjugation | - | Glycine, taurine | Bile acids, benzoic acid |
Important Notes on Phase II
- Phase II does not always follow Phase I - e.g., isoniazid undergoes N-acetylation (Phase II) first, then the acetyl-isoniazid undergoes hydrolysis (Phase I)
- Some conjugation reactions produce reactive/toxic intermediates rather than detoxifying: acyl glucuronidation of NSAIDs, O-sulfation of N-hydroxyacetylaminofluorene, N-acetylation of isoniazid can all generate hepatotoxic species
- Sulfation can activate prodrugs: Minoxidil sulfate is the active vasodilator form
- Morphine-6-glucuronide is more potent than morphine itself
4. Enzyme Induction and Inhibition
Enzyme Induction
Induction increases the quantity/activity of CYP enzymes - most relevant for drugs with low-to-moderate hepatic extraction fractions (capacity-dependent drugs).
Two classical types (Tietz Textbook of Laboratory Medicine, 7th Ed.):
- Phenobarbital-type: Broad induction - increases CYP450, CYP450 reductase, liver weight, hepatic blood flow; primarily induces CYP2D6 (debrisoquine substrate)
- 3-Methylcholanthrene/polycyclic aromatic hydrocarbon-type (tobacco smoke): Induces CYP1A, increases theophylline clearance without affecting antipyrine
Enzyme Inhibition
Mechanisms include:
- Substrate competition
- Competitive or non-competitive inhibition
- Product inhibition
- Enzyme repression (decreased synthesis or increased degradation)
Classic inhibitors: cimetidine, chloramphenicol, valproic acid, allopurinol, erythromycin, fluconazole, quinidine
5. Hepatic Extraction and Clearance
Hepatic clearance = Liver blood flow × Extraction ratio (Tietz Lab Medicine, 7th Ed.; Morgan & Mikhail's Clinical Anesthesiology, 7th Ed.)
| Type | Extraction Ratio | Rate-Limiting Factor | Effect of Enzyme Induction | Examples |
|---|---|---|---|---|
| Flow-dependent | High (>70%) | Liver blood flow | Minimal | Propofol, propranolol, lidocaine, morphine, nitroglycerin |
| Capacity-dependent | Low (<30%) | Metabolic capacity | Significant | Methadone (10%), alfentanil (15%), diazepam, warfarin |
- High-extraction drugs: cirrhosis has little effect; reducing hepatic blood flow (e.g., β-blockers, heart failure) reduces clearance
- Low-extraction drugs: liver damage or enzyme inhibition significantly reduces clearance; enzyme induction significantly increases it
6. Pharmacogenomics of Biotransformation
Genetic polymorphisms markedly alter biotransformation:
- CYP2D6: Poor metabolizers (PMs) - codeine provides no analgesia; ultra-rapid metabolizers (UMs) - risk of opioid toxicity from codeine
- CYP2C19: PMs - omeprazole accumulates (better H. pylori eradication); clopidogrel fails as antiplatelet (requires CYP2C19 activation)
- NAT2: Slow acetylators - isoniazid, hydralazine, procainamide toxicity (peripheral neuropathy, lupus-like syndrome)
- TPMT: Poor methylators - 6-mercaptopurine/azathioprine myelotoxicity
- CYP3A5: ~50% of African Americans express CYP3A5; these individuals need higher tacrolimus doses than Caucasians
7. Toxicology: Biotransformation to Toxic Products
Biotransformation is not always detoxifying. The classic example is acetaminophen hepatotoxicity (Katzung's Basic & Clinical Pharmacology, 16th Ed.):
- Therapeutic doses: 95% metabolized via glucuronidation + sulfation (safe); 5% via CYP2E1/3A4 → NAPQI (reactive), immediately conjugated by GSH
- Overdose: glucuronidation and sulfation pathways saturate; CYP pathway dominates → NAPQI accumulates → GSH depleted → NAPQI attacks hepatocellular proteins + redox cycling → oxidative stress → centrilobular necrosis
- Treatment: N-acetylcysteine (NAC) replenishes GSH
Other examples of toxicogenic biotransformation:
- INH → isonicotinic acyl radical → attacks P450 proteins → immune-mediated hepatotoxicity
- Cyclophosphamide → acrolein (bladder toxicity) via CYP2B6
- Benzene → benzene epoxide → aplastic anemia
Summary
| Feature | Phase I | Phase II |
|---|---|---|
| Reactions | Oxidation, reduction, hydrolysis | Conjugation (glucuronidation, sulfation, acetylation, etc.) |
| Main enzymes | CYP450 system, FMOs | UGTs, SULTs, NATs, GSTs |
| Product polarity | Increased | Highly polar (water-soluble) |
| Result | Modified/inactive metabolite | Usually inactive conjugate (exceptions exist) |
| Location | ER (microsomes) | Cytosol + microsomes |
| Speed | Slower | Faster |
Sources:
- Katzung's Basic and Clinical Pharmacology, 16th Edition, Ch. 4
- Tietz Textbook of Laboratory Medicine, 7th Edition, Ch. on Biotransformation
- Morgan & Mikhail's Clinical Anesthesiology, 7th Edition, Ch. on Pharmacokinetics
- Yamada's Textbook of Gastroenterology, 7th Edition, Ch. 28 (Drug Metabolism)
In short
Biotransformation - Quick Summary
Definition: The body's chemical modification of drugs, converting lipophilic molecules into water-soluble forms for excretion.
Primary site: Liver (also GI tract, lungs, kidneys, plasma)
Phase I - "Functionalization"
- Reactions: Oxidation, reduction, hydrolysis
- Main enzyme: Cytochrome P450 (CYP) system
- Result: Adds/unmasks a polar group (-OH, -NH₂, -SH)
- Products may be inactive, active, or toxic
Phase II - "Conjugation"
- Attaches an endogenous molecule to the Phase I product (or directly to the drug)
- Types: Glucuronidation, sulfation, acetylation, methylation, glutathione conjugation
- Result: Highly water-soluble conjugate → excreted in urine/bile
- Generally faster than Phase I
Key Points
| Concept | Key Fact |
|---|---|
| First-pass effect | Oral drugs metabolized by liver before reaching systemic circulation |
| Enzyme induction | ↑ CYP activity (e.g., rifampin, phenobarbital) → ↓ drug levels |
| Enzyme inhibition | ↓ CYP activity (e.g., ketoconazole, ciprofloxacin) → ↑ drug levels |
| High extraction drugs | Clearance depends on liver blood flow (e.g., propofol, lidocaine) |
| Low extraction drugs | Clearance depends on enzyme capacity (e.g., warfarin, diazepam) |
| Prodrugs | Activated by biotransformation (e.g., codeine → morphine, clopidogrel → active thiol) |
| Toxic metabolites | Acetaminophen overdose → NAPQI → hepatotoxicity when GSH depleted |
Bottom line: Phase I modifies the drug; Phase II packages it for elimination. CYP3A4 handles ~30% of all drugs. Genetic polymorphisms and drug interactions at CYP enzymes are major sources of variability in drug response.
Factors modifying drug action
Finding Sources
Finding Sources
Finding Sources
Finding Sources
This is a shared conversation. Sign in to Orris to start your own chat.