wilson's disease defination, epidemiology, etiology, risk factors, complications, diagnosis, pathophysiology, non pharmacological therapy and pharmacological therapy
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Wilson disease"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Searching Images
Wilson disease Kayser-Fleischer rings copper deposits liver pathology
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive, high-quality data from multiple authoritative textbooks. Let me compile the full Wilson's disease answer:
Wilson's Disease — Comprehensive Clinical Overview
1. Definition
Wilson disease (hepatolenticular degeneration) is an autosomal recessive disorder of copper transport caused by loss-of-function mutations in both alleles of the ATP7B gene, which encodes a copper-transporting ATPase expressed primarily in hepatocytes. The consequence is impaired excretion of hepatic copper into bile and reduced incorporation of copper into ceruloplasmin, leading to toxic accumulation of copper principally in the liver, brain (especially the basal ganglia), and corneas. First described in 1912 by neurologist S.A.K. Wilson, it is progressive if untreated but highly responsive to therapy when diagnosed early.
- Goldman-Cecil Medicine, p. 2325 | Harrison's Principles of Internal Medicine 22E, p. 3382
2. Epidemiology
| Parameter | Value |
|---|---|
| Incidence | ~1 in 30,000–40,000 live births |
| Heterozygous carrier prevalence | 1 in 90–180 in the general population |
| Age of onset | Typically 6–40 years (reported up to 5th–6th decade) |
| Sex | Equal male and female distribution |
| Population-based genomic estimates | As high as 1 in 7,000 (suggesting under-diagnosis) |
Incidence is higher in populations where consanguinity is common. The disparity between population-based genomic prevalence estimates (~1 in 7,000) and clinical estimates (~1 in 30,000) may reflect incomplete penetrance or missed diagnoses. Advances in whole-genome sequencing from newborn dried blood spots may transform presymptomatic screening.
- Goldman-Cecil Medicine, p. 2325 | Harrison's, p. 3383
3. Etiology
Wilson disease is caused exclusively by biallelic loss-of-function mutations in ATP7B on chromosome 13q14.3. Key molecular points:
-
650 pathogenic or likely pathogenic variants are catalogued in major ATP7B databases
-
Most patients are compound heterozygotes (different mutations on each allele)
-
Common variants include H1069Q (common in Europeans), M645R, and R778L (common in East Asians)
-
Large deletions are relatively uncommon; most pathogenic variants (~97%) are in coding exons or intronic sequences
-
Related gene ATP7A (mutated in Menkes disease) is X-linked; ATP7B is autosomal
-
Modifier genes (e.g., CAT, SOD2, MTHFR) may influence clinical expression in some individuals
-
Harrison's, p. 3383 | Goldman-Cecil, p. 2325
4. Risk Factors
- Family history: First-degree relatives of an index case have a 1 in 4 risk (autosomal recessive); siblings must be screened
- Consanguinity: Increases risk of homozygous mutations
- Dietary copper excess: High-copper foods (shellfish, liver, nuts, chocolate, mushrooms) can precipitate or accelerate manifestations in affected individuals
- Drinking water copper: Concentrations approaching 1.3 mg/L from copper pipes can worsen copper loading
- Non-compliance with treatment: Leads to symptomatic relapse and liver failure even in previously well-controlled patients
5. Pathophysiology
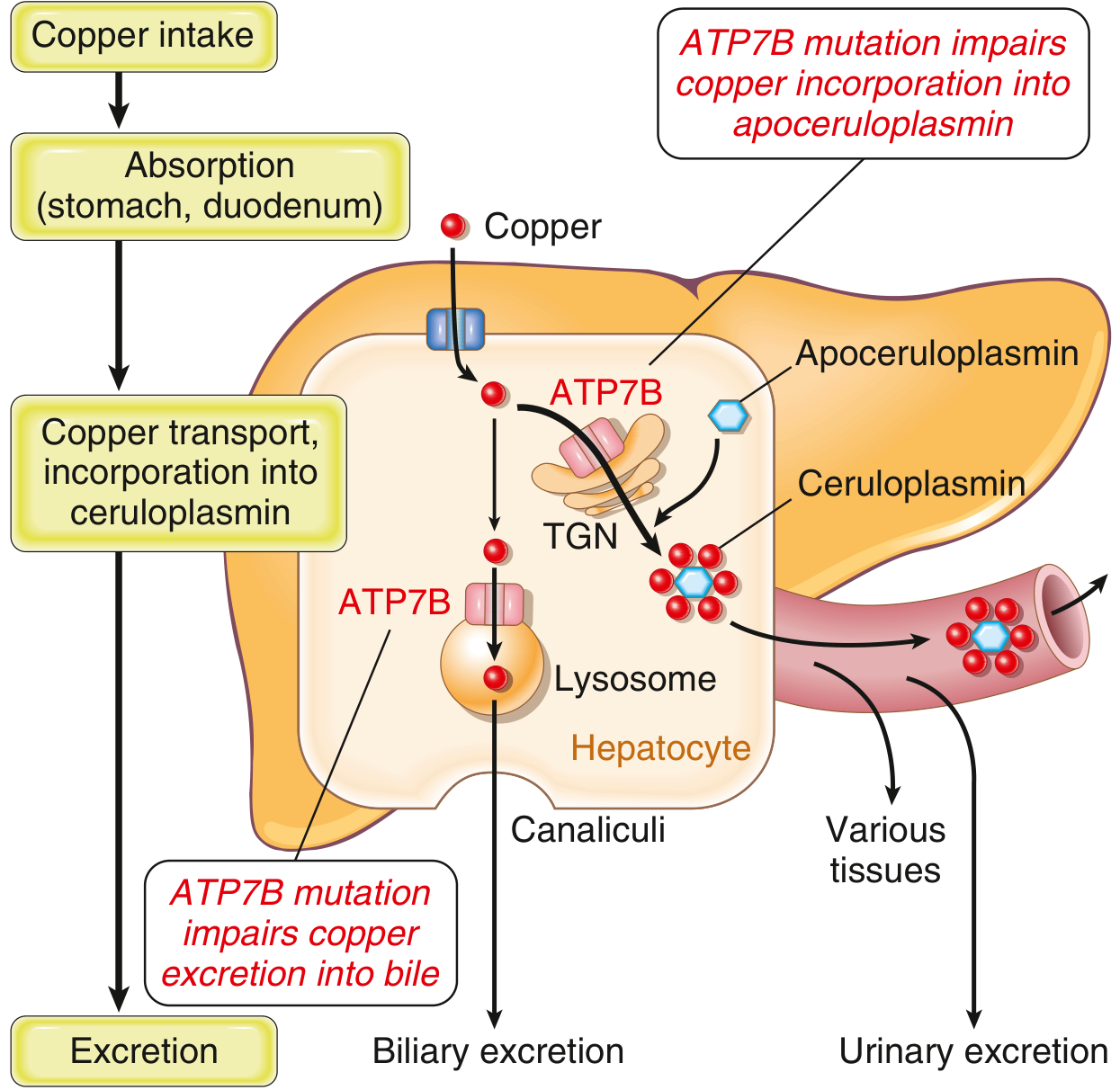
Normal copper homeostasis:
- 1–3 mg dietary copper consumed daily; ~50% absorbed in stomach and duodenum via hCTR1 transporter
- Copper carried by albumin/amino acids to the liver via portal blood
- In hepatocytes, copper is: (1) incorporated into ceruloplasmin via ATP7B in the trans-Golgi network; (2) used in copper-dependent enzymes; (3) stored bound to metallothionein; or (4) excreted into bile via ATP7B in lysosomes
- Biliary excretion is the major route of copper elimination — bile copper does not undergo enterohepatic recirculation
In Wilson disease (ATP7B loss of function):
- Biliary copper excretion fails → hepatic copper accumulates
- Copper accumulates in hepatocyte cytoplasm and lysosomes → increased ROS production → hepatocyte injury (steatosis → acute hepatitis → chronic hepatitis → cirrhosis)
- With progressive liver damage, non-ceruloplasmin-bound copper is released into circulation → hemolytic anemia, and copper deposition in:
- Brain (basal ganglia — caudate, putamen; also midbrain, pons, thalamus, cerebellum) → neuropsychiatric disease
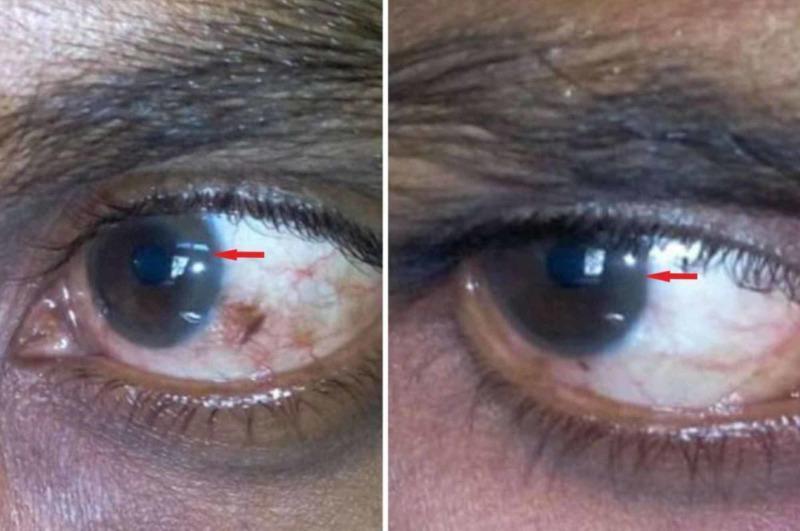
- Cornea (Descemet membrane) → Kayser-Fleischer rings
- Kidney (proximal tubules) → Fanconi syndrome
- Bones, joints, parathyroid glands
- Although serum ceruloplasmin is low (impaired copper loading of apoceruloplasmin → unstable apoprotein), low ceruloplasmin plays no role in the pathogenesis of disease; copper itself drives toxicity
- Robbins Basic Pathology, p. [block 6] | Goldman-Cecil, p. 2325–2326
6. Complications
Hepatic
- Steatosis (fatty liver) — earliest change
- Acute hepatitis (can mimic viral hepatitis)
- Chronic active hepatitis with fibrosis
- Cirrhosis — present in most patients with neurological disease
- Acute liver failure (ALF) — 5% of patients; presents with Coombs-negative hemolytic anemia, coagulopathy, markedly elevated bilirubin; alkaline phosphatase:bilirubin ratio <4 and AST:ALT ratio >2.2 are diagnostic clues
Neurological
- Dysarthria, dystonia, rigidity
- Tremor ("wing-beating" or postural/resting)
- Choreiform movements, ataxia, gait disturbance
- Dysphagia, drooling, "sardonic" smile (risus sardonicus)
- Seizures (minority of patients)
- MRI: abnormal T2/FLAIR signal in putamen, midbrain, pons, thalamus, cerebellum; cerebral atrophy
Psychiatric
- Personality and mood changes (most common — ~50% of patients)
- Depression (~30%)
- Bipolar spectrum symptoms (~20%)
- Suicidal ideation (5–15%)
- Psychosis, anxiety, irritability, aggression
- Cognitive decline (frontosubcortical pattern)
Ophthalmic
- Kayser-Fleischer rings: golden-brown deposits of copper in Descemet membrane at corneal limbus — present in ~95% of patients with neurological disease; ~50–65% with hepatic-only presentation; slit-lamp required for early detection
- Sunflower cataracts: copper in the lens (rare)
Hematological
- Coombs-negative hemolytic anemia: direct toxicity of copper on RBC membranes; can be catastrophic in ALF
Renal
- Renal tubular Fanconi syndrome: aminoaciduria, glycosuria (normoglycemic), phosphaturia, uricosuria, calciuria
- Nephrolithiasis, hypercalciuria
Skeletal
- Osteoporosis, osteomalacia, rickets (from renal calcium/phosphorus loss)
- Osteoarthritis (knees and wrists — from copper in bone/cartilage)
Reproductive
- Secondary amenorrhea, delayed puberty (from liver disease)
- Increased spontaneous miscarriage in untreated pregnant women
7. Diagnosis
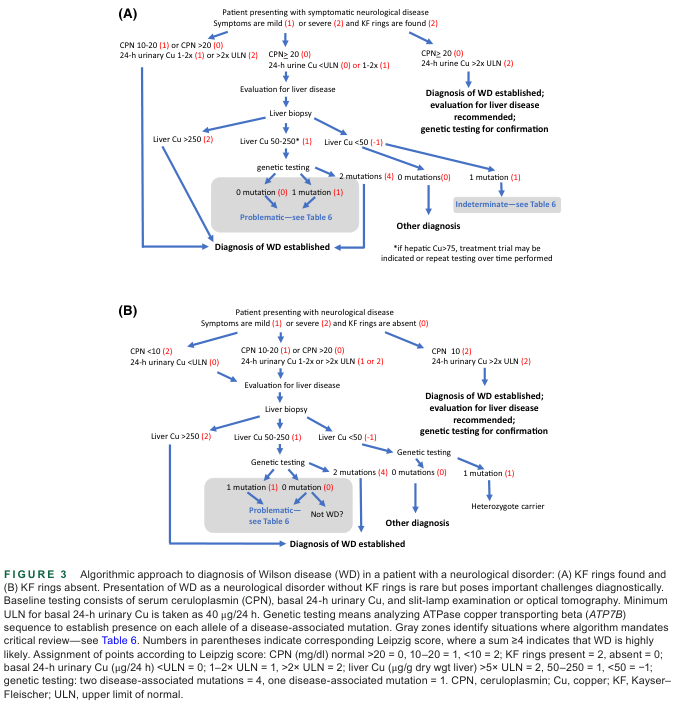
Diagnosis combines clinical, biochemical, and molecular data, often using the Leipzig Scoring System (endorsed by EASL).
Leipzig Scoring System (points)
| Finding | Points |
|---|---|
| Kayser-Fleischer rings present | +2 |
| Neuropsychiatric symptoms | +2 |
| Serum ceruloplasmin <11.5 mg/dL | +2 |
| Serum ceruloplasmin 11.5–20 mg/dL | +1 |
| 24-h urine copper >2× ULN or >100 μg/day | +2 |
| 24-h urine copper 1–2× ULN | +1 |
| Liver copper >250 μg/g dry weight | +2 |
| Liver copper 50–250 μg/g dry weight | +1 |
| Liver copper <50 μg/g dry weight | −1 |
| Two ATP7B mutations identified | +4 |
| One ATP7B mutation identified | +1 |
| Coombs-negative hemolytic anemia | +1 |
Score ≥4: Wilson disease likely; Score 2–3: possible, further testing needed; Score ≤1: diagnosis unlikely.
Key Investigations
| Test | Finding in Wilson Disease | Notes |
|---|---|---|
| Serum ceruloplasmin | <20 mg/dL (usually <11.5 mg/dL) | Most efficient initial test; low in 95% |
| 24-h urine copper | >100 μg/day | Use acid-washed collection containers |
| Serum copper (total) | Low | Paradoxically low despite copper accumulation |
| Liver biopsy — quantitative copper | >200 μg/g dry weight (gold standard) | By ICP-mass spectrometry or atomic absorption |
| Slit-lamp examination | Kayser-Fleischer rings | Essential; OCT of anterior segment increasingly useful |
| Serum alkaline phosphatase/bilirubin | <4 in Wilson ALF | Distinguishes from other causes of ALF |
| AST/ALT ratio | >2.2 in Wilson ALF | |
| Genetic testing (ATP7B) | Biallelic mutations | Confirms diagnosis; guides family screening |
| MRI brain | T2/FLAIR hyperintensity in basal ganglia, brainstem | Indicates neurological involvement |
| Penicillamine challenge test | >5-fold increase in urine copper after 500 mg oral penicillamine | Useful in children with equivocal urine copper |
- Harrison's, p. 3382–3383 | Goldman-Cecil, p. 2326 | Bradley and Daroff's Neurology
8. Non-Pharmacological Therapy
Dietary Copper Restriction
- Avoid high-copper foods: shellfish (especially oysters, crab), liver and organ meats, mushrooms, chocolate, nuts, seeds, dried fruit
- Test drinking water: avoid if copper concentration approaches 1.3 mg/L (common from copper plumbing)
- Dietary restriction alone is insufficient but is an important adjunct
Rehabilitation Therapy
- Speech therapy: for dysarthria and dysphagia
- Physical therapy: for tremor, dystonia, gait disturbance, and motor rehabilitation
- Occupational therapy: for handwriting difficulties and activities of daily living
- These are particularly important in newly diagnosed patients with neurological manifestations
Psychological Support
- Neuropsychiatric symptoms are present in up to 70% of patients long-term; psychological evaluation and counseling are integral to management
- Management of depression, anxiety, personality disorders — with specific caution regarding neuroleptic sensitivity
Family Screening and Genetic Counseling
- All first-degree relatives of a diagnosed patient must be screened (serum ceruloplasmin, slit-lamp, urine copper, genetic testing)
- Presymptomatic treatment of affected relatives prevents disease manifestation
Liver Transplantation (Non-pharmacological surgical)
- Indications: ALF unresponsive to medical therapy; end-stage irreversible liver disease; rarely, severe intractable neurological disease
- Outcome: Cures Wilson disease — corrects the metabolic defect, Kayser-Fleischer rings fade, neurological function often improves; disease does not recur
- 5% of patients with ALF require urgent transplantation for survival
9. Pharmacological Therapy
Treatment is lifelong. Non-compliance inevitably leads to symptomatic recurrence and potentially fatal liver damage.
Copper Chelators (First-line for symptomatic patients)
D-Penicillamine (Cuprimine)
- Mechanism: Free thiol binds free copper → dramatically increases urinary copper excretion; does not correct the basic biliary excretion defect
- Dose: Start at 250 mg twice daily; titrate to 15–20 mg/kg/day in 2–4 divided doses
- Pyridoxine (vitamin B6) co-administration: Mandatory to prevent B6 deficiency during chronic use
- Side effects (~20% intolerance):
- Early: hypersensitivity reactions (rash, fever, lymphadenopathy)
- Late: nephrotoxicity (membranous glomerulonephritis), hematologic toxicity (thrombocytopenia, leukopenia)
- Dermatologic: elastosis perforans serpiginosa (distinctive serpiginous rash at neck/axillae)
- Neurological worsening: 20–50% of neurologically presenting patients experience paradoxical deterioration, some permanent — a major clinical concern
- Monitoring: 24-h urine copper, CBC, urinalysis
Trientine (Triethylenetetramine dihydrochloride — Syprine)
- Mechanism: Chelates copper → enhances urinary copper excretion
- Indication: First-line alternative in patients intolerant of penicillamine; preferred in neurologically presenting patients (lower risk of neurological worsening); noninferior to D-penicillamine for copper maintenance therapy
- Advantages: Better side-effect profile than penicillamine
- Trientine tetrahydrochloride (newer formulation) — demonstrated non-inferiority to D-penicillamine in stable Wilson disease
Tetrathiomolybdate (TM) / Tetrahydroxytetramolybdate (bis-choline tetrathiomolybdate)
- Mechanism: Forms stable tripartite complexes with albumin and copper → reduces both GI copper absorption and circulating free copper; promotes biliary copper excretion
- Advantages: Fast-acting — restores normal copper balance within weeks (vs. months for other agents); may be especially appropriate as initial therapy in neurologically presenting patients due to lower risk of neurological worsening
- Status: Experimental/emerging; phase III trials ongoing
Zinc Salts (Maintenance / Presymptomatic)
Zinc Acetate (Galzin) / Zinc Gluconate / Zinc Sulfate
- Mechanism: Induces metallothionein synthesis in intestinal epithelial cells → metallothionein binds dietary copper in enterocytes → blocked absorption → copper excreted with sloughed enterocytes in feces (does not enhance urinary excretion)
- Indications:
- Presymptomatic patients (first-line)
- Maintenance therapy after initial "de-coppering" with chelators
- Pregnancy (preferred due to safety profile; teratogenic risk of chelators)
- Dose: 150 mg elemental zinc/day in 3 divided doses (taken between meals)
- Advantages: Minimal systemic toxicity; safe in pregnancy
- Disadvantages:
- Slow onset — 4–6 months to restore copper balance when used as initial monotherapy
- 10–20% experience dyspepsia
- Higher incidence of hepatic decompensation with long-term use vs. chelation therapy
- Not recommended as sole initial therapy in symptomatic patients due to slow action
Treatment in Special Situations
| Situation | Preferred Approach |
|---|---|
| Initial symptomatic hepatic disease | D-penicillamine or trientine |
| Initial neurological disease | Trientine or tetrathiomolybdate (avoid penicillamine if possible) |
| Presymptomatic | Zinc acetate monotherapy |
| Pregnancy | Zinc (maintain at usual dose); reduce chelator dose if on chelation; no breastfeeding |
| Acute liver failure | Bridge to liver transplantation; chelation may not be sufficient |
| Maintenance after de-coppering | Zinc or low-dose chelator |
| Tremor/dystonia (symptomatic) | Specific anti-tremor/anti-dystonia agents alongside copper treatment |
Emerging Therapies
- Gene therapy: Adeno-associated virus (AAV)-mediated ATP7B introduction into hepatocytes — Phase I/II clinical trials ongoing; transduction of only 20% of hepatocytes normalized copper homeostasis in animal models
- Methanobactin: Bacterial peptide with high copper affinity, traverses mitochondrial membranes — preclinical evidence in rat model of advanced Wilson disease
Monitoring of Treatment
- 24-h urine copper: should remain 200–500 μg/day (not zero — over-chelation harmful)
- Serum ceruloplasmin and copper (non-ceruloplasmin-bound "free" copper)
- Liver function tests and CBC (for penicillamine toxicity)
- Slit-lamp exam: Kayser-Fleischer rings fade with effective treatment
- MRI brain: may show partial improvement with sustained treatment
Summary Table
| Domain | Key Points |
|---|---|
| Definition | Autosomal recessive ATP7B mutation → impaired biliary copper excretion → copper toxicity |
| Epidemiology | 1:30,000 births; carrier frequency 1:90–180; onset 6–40 years |
| Etiology | >650 ATP7B mutations; compound heterozygotes most common |
| Pathophysiology | Hepatic copper accumulation → ROS → hepatocyte damage → overflow to brain, cornea, kidney |
| Hallmark sign | Kayser-Fleischer rings (95% of neurological cases) |
| Diagnosis | Leipzig score ≥4; ceruloplasmin <11.5; urine Cu >100 μg/24h; liver Cu >200 μg/g |
| Non-pharm Rx | Low-copper diet, water testing, PT/OT/speech therapy, liver transplant for ALF/end-stage |
| Chelators | D-penicillamine (first-line), trientine (neurological/intolerance), tetrathiomolybdate (emerging) |
| Zinc | Maintenance/presymptomatic/pregnancy; slow onset; do not use as initial monotherapy |
| Prognosis | Excellent if treated early; lifelong therapy mandatory; 3× higher mortality than general population if undertreated |
Sources:
- Goldman-Cecil Medicine International Edition, Chapter 195 (Wilson Disease)
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 427 (Wilson's Disease)
- Robbins & Kumar Basic Pathology, Chapter 14 (Wilson Disease)
- Bradley and Daroff's Neurology in Clinical Practice, Chapter on Wilson Disease
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease, Chapter on Wilson Disease
Lymphadenitis defination, epidemiology, etiology, risk factors, complications, diagnosis, pathophysiology, non pharmacological therapy and pharmacological therapy
Reading File
Finding Sources
Finding Sources
Searching Images
lymphadenitis lymph node histology inflammation pathology
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
You've run out of free credits. Sign up for a free account to continue using Orris.
This is a shared conversation. Sign in to Orris to start your own chat.