Comprehensive and detailed information onIntegrated metabolism between carbohydrates proteins and lipids Include all the cycles and interconnection and along with clinical correlations Be precise and clear as I am preparing for neetpg usmle inicet
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive material from authoritative textbooks. Let me compile the complete, exam-focused answer.
Integrated Metabolism: Carbohydrates, Proteins, and Lipids
THE CORE CONCEPT
All three macronutrient pathways converge at a few common metabolic hubs. Understanding these hubs and the cycles connecting them is the foundation of integrated metabolism.
Key hubs:
- Pyruvate - crossroads of carbohydrate, amino acid, and lactate metabolism
- Acetyl-CoA - final common pathway entry into the TCA cycle; also precursor for ketone bodies and fatty acid synthesis
- Oxaloacetate (OAA) - TCA cycle intermediate; also a gluconeogenic substrate
- Alpha-ketoglutarate (alpha-KG) - links amino acid catabolism to TCA cycle
- Glucose-6-phosphate - branch point for glycolysis, glycogen synthesis, and pentose phosphate pathway
SECTION 1: THE TCA (KREBS/CITRIC ACID) CYCLE - THE CENTRAL HUB
The TCA cycle does not just oxidize acetyl-CoA. It is the point where carbon skeletons from ALL three macronutrients converge.
Entry points into TCA cycle
| Macronutrient | Metabolite | Entry Point in TCA |
|---|---|---|
| Carbohydrates | Pyruvate -> Acetyl-CoA | Combines with OAA -> Citrate |
| Lipids | Fatty acid beta-oxidation -> Acetyl-CoA | Same as above |
| Proteins - glucogenic AA | Alanine, Serine, Cysteine -> Pyruvate | Via Acetyl-CoA or direct |
| Proteins - glucogenic AA | Aspartate, Asparagine -> OAA | Directly into TCA |
| Proteins - glucogenic AA | Glutamate, Glutamine, Pro, His, Arg -> alpha-KG | Into TCA |
| Proteins - glucogenic AA | Isoleucine, Methionine, Val, Thr -> Succinyl-CoA | Into TCA |
| Proteins - ketogenic AA | Leucine, Lysine -> Acetyl-CoA | Into TCA as ketogenic |
| Mixed (glucogenic + ketogenic) | Ile, Phe, Trp, Tyr, Thr | Multiple entry points |
Exam tip (NEET PG/INICET/USMLE): Leucine and Lysine are the ONLY purely ketogenic amino acids (no net glucose production). All others are either purely glucogenic or mixed.
SECTION 2: THE MAJOR INTERCONNECTING CYCLES
2A. THE CORI CYCLE (Lactic Acid Cycle)
Organs involved: Skeletal muscle / RBCs <--> Liver
Mechanism:
- Muscle and RBCs metabolize glucose via glycolysis -> lactate (anaerobic; RBCs have no mitochondria)
- Lactate is exported into blood
- Liver takes up lactate -> converts to pyruvate (lactate dehydrogenase)
- Pyruvate enters gluconeogenesis -> glucose
- Glucose re-enters blood for use by muscle/RBCs
Energy economics:
- Muscle generates 2 ATP from glucose -> lactate
- Liver uses 6 ATP to convert lactate -> glucose (gluconeogenesis)
- Net: energy transfer from liver to muscle (liver subsidizes anaerobic muscle work)
- Neither cycle yields NEW carbon skeletons - carbon is recycled
Key fact for exams: The Cori cycle does NOT produce new net glucose from outside sources. It recycles existing carbon. The ATP cost in the liver comes from fatty acid oxidation.
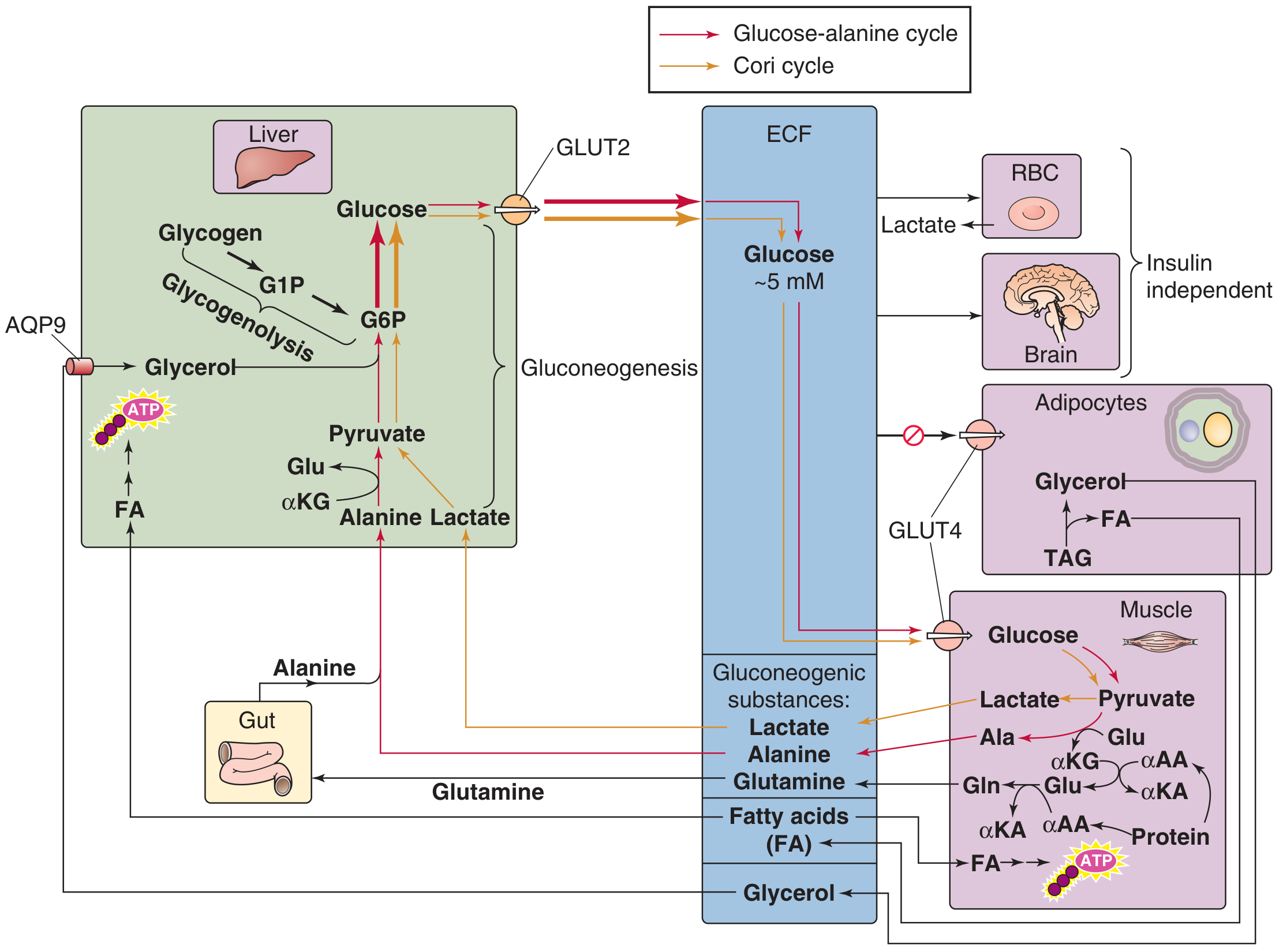
2B. THE GLUCOSE-ALANINE CYCLE (Cahill Cycle)
Organs involved: Skeletal muscle <--> Liver
Mechanism:
- Muscle proteins are catabolized during fasting -> amino acids
- Amino groups transferred to pyruvate (from glycolysis) via alanine aminotransferase (ALT/SGPT) -> alanine + alpha-ketoglutarate
- Alanine exported from muscle into blood
- Liver takes up alanine -> transamination back to pyruvate + glutamate
- Pyruvate -> gluconeogenesis -> glucose (released back to blood)
- Glutamate -> urea cycle -> urea excreted by kidney
Dual purpose of glucose-alanine cycle:
- Provides gluconeogenic substrate to liver (carbon)
- Transfers nitrogen from muscle to liver for urea synthesis (nitrogen detoxification)
- Uses alanine as a non-toxic nitrogen carrier (unlike free ammonia)
Key difference from Cori cycle:
- Cori cycle: energy transfer only
- Glucose-alanine cycle: energy transfer + nitrogen transfer
Exam memory hook: ALT (alanine aminotransferase) is elevated in liver disease - this is the enzyme that converts alanine to pyruvate (and vice versa) in the glucose-alanine cycle. ALT is a liver-specific marker because the liver is where this transamination is most active.
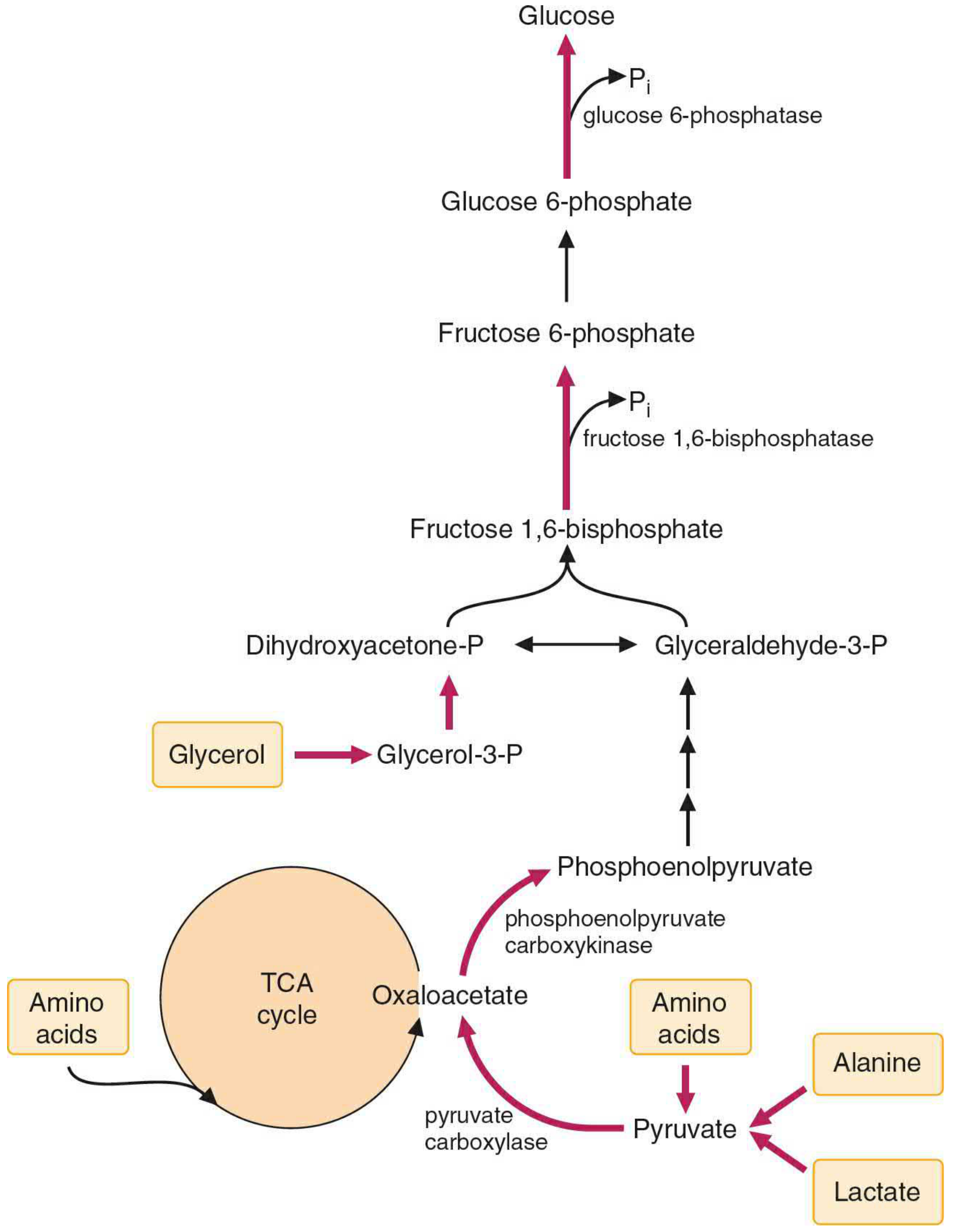
2C. GLUCONEOGENESIS - THE MASTER INTEGRATION POINT
Gluconeogenesis is where all three macronutrients donate carbons to make glucose:
| Precursor | Source Macronutrient | Entry Step |
|---|---|---|
| Lactate | Carbohydrate (anaerobic glycolysis) | -> Pyruvate -> OAA -> PEP |
| Alanine | Protein (muscle catabolism) | -> Pyruvate -> OAA -> PEP |
| Glutamine | Protein | -> alpha-KG -> OAA -> PEP |
| Glycerol | Lipid (triglyceride hydrolysis) | -> DHAP -> F1,6-BP |
| Propionate | Odd-chain fatty acids | -> Succinyl-CoA -> OAA |
| Oxaloacetate | TCA intermediates (from AA) | -> PEP directly |
The 3 irreversible steps of glycolysis that are BYPASSED in gluconeogenesis:
| Glycolysis (irreversible) | Gluconeogenesis bypass | Enzyme |
|---|---|---|
| Pyruvate kinase: PEP -> Pyruvate | Pyruvate -> OAA -> PEP | Pyruvate carboxylase + PEPCK |
| PFK-1: F6P -> F1,6-BP | F1,6-BP -> F6P | Fructose-1,6-bisphosphatase |
| Hexokinase/Glucokinase: Glucose -> G6P | G6P -> Glucose | Glucose-6-phosphatase (liver/kidney only!) |
USMLE/NEET PG key: Von Gierke disease = glucose-6-phosphatase deficiency -> cannot release free glucose from liver -> severe fasting hypoglycemia + hepatomegaly + lactic acidosis.
SECTION 3: LIPID-CARBOHYDRATE INTEGRATION
Fatty Acid Synthesis from Carbohydrates
In the fed state with excess glucose:
- Glucose -> Glycolysis -> Pyruvate
- Pyruvate -> Acetyl-CoA (pyruvate dehydrogenase, mitochondrial)
- Acetyl-CoA condenses with OAA -> Citrate
- Citrate shuttle: Citrate exits mitochondria -> cytoplasm
- ATP-citrate lyase cleaves citrate -> Acetyl-CoA (cytoplasm) + OAA
- Cytoplasmic Acetyl-CoA is used for fatty acid synthesis (acetyl-CoA carboxylase -> malonyl-CoA -> palmitate)
Why can't we convert fat -> glucose net?
- Acetyl-CoA (from beta-oxidation of even-chain fatty acids) CANNOT be converted to pyruvate or OAA NET
- Acetyl-CoA enters TCA but the 2 carbons added are lost as 2 CO2 per turn
- Therefore: fat cannot contribute net carbons to gluconeogenesis (except glycerol and odd-chain fatty acids)
- Glyoxylate cycle allows this in plants/bacteria/yeast (isocitrate lyase + malate synthase) but HUMANS LACK THIS CYCLE
High-yield exam point: This is why prolonged starvation/DKA leads to muscle protein catabolism - fat alone cannot maintain glucose levels for the brain, so protein must be broken down.
Ketone Body Formation (Ketogenesis)
In fasting/starvation/DKA:
- Lipolysis in adipocytes -> FFAs released into blood
- Liver beta-oxidation: FFAs -> Acetyl-CoA
- With low carbohydrate intake: OAA is depleted (pulled into gluconeogenesis)
- Acetyl-CoA cannot enter TCA (no OAA partner) -> accumulates
- 2 Acetyl-CoA -> Acetoacetyl-CoA -> HMG-CoA (HMG-CoA synthase) -> Acetoacetate + beta-hydroxybutyrate + Acetone
- Ketone bodies exported to brain, heart, muscle as alternative fuel
Why does OAA fall in starvation?
- Glucagon activates PEPCK -> OAA -> PEP (for gluconeogenesis)
- OAA is depleted from TCA, leaving acetyl-CoA "stranded"
- This is the biochemical basis of ketosis
Regulation: HMG-CoA synthase (mitochondrial) is the rate-limiting step of ketogenesis. Malonyl-CoA (the first product of fatty acid synthesis) INHIBITS CPT-1, preventing fatty acid entry into mitochondria - so when fed state is active (FA synthesis on), ketogenesis is off.
SECTION 4: PROTEIN-LIPID INTEGRATION
Lipogenic amino acids
- Glucogenic amino acids -> glucose -> can be stored as glycogen or converted to fat via Acetyl-CoA
- Ketogenic amino acids -> Acetyl-CoA -> directly enter FA synthesis or ketogenesis
Cholesterol synthesis
- Acetyl-CoA (from all three macronutrients) -> HMG-CoA (cytoplasmic) -> Mevalonate -> Cholesterol
- Rate-limiting enzyme: HMG-CoA reductase (target of statins)
- HMG-CoA has two fates:
- Cytoplasmic HMG-CoA -> cholesterol (via HMG-CoA reductase)
- Mitochondrial HMG-CoA -> ketone bodies (via HMG-CoA lyase)
Amino acids as lipid precursors
- Serine -> Phospholipids (phosphatidylserine)
- Glycine + Succinyl-CoA -> Heme synthesis
- Methionine -> SAM -> methylation of lipids (phosphatidylcholine)
SECTION 5: THE UREA CYCLE AND ITS INTEGRATION
The urea cycle is the mechanism for nitrogen disposal from amino acid catabolism. It is intimately linked to the TCA cycle via the aspartate-argininosuccinate shunt (also called the "Krebs bicycle"):
- Aspartate (from OAA + glutamate via transamination) donates a nitrogen to argininosuccinate in the urea cycle
- Fumarate is released from argininosuccinate -> enters TCA cycle directly
- TCA: Fumarate -> Malate -> OAA
- OAA + glutamate -> Aspartate + alpha-KG (regenerating aspartate for next urea cycle turn)
This means the urea cycle and TCA cycle share intermediates and run in a coordinated fashion - the "bicycle" concept.
SECTION 6: METABOLIC STATE-BASED INTEGRATION
Fed State (Post-prandial, Insulin dominant)
| Tissue | Key Activity |
|---|---|
| Liver | Glycolysis, glycogen synthesis, fatty acid synthesis (de novo lipogenesis), VLDL secretion |
| Adipose | Glucose uptake (GLUT4), LPL active, triglyceride storage, inhibits lipolysis |
| Muscle | Glucose uptake (GLUT4), glycogen synthesis, protein synthesis |
| Brain | Glucose uptake (GLUT3, insulin-independent), glucose oxidation |
- Key enzyme activated: Acetyl-CoA carboxylase (FA synthesis), glycogen synthase, pyruvate kinase
- Key enzyme inhibited: Hormone-sensitive lipase, gluconeogenesis enzymes
Fasting State (Post-absorptive, Glucagon dominant)
| Tissue | Key Activity |
|---|---|
| Liver | Glycogenolysis (first 6-12 hr), then gluconeogenesis; ketogenesis |
| Adipose | Lipolysis (HSL activated by glucagon/epinephrine) -> FFAs + glycerol |
| Muscle | Glucose uptake falls; uses FFAs, ketones; protein catabolism -> alanine/glutamine export |
| Brain | Initially glucose; after 3-4 days of starvation, adapts to use ketone bodies (up to 75% of energy) |
| Kidney | Gluconeogenesis from glutamine (significant during prolonged fasting) |
Prolonged Starvation
- 0-4 hr: Glycogenolysis
- 4-16 hr: Gluconeogenesis from lactate, alanine, glycerol
- 16 hr - 2 days: Gluconeogenesis from muscle protein (major); fat oxidation + ketogenesis intensifies
- 3-7 days onwards: Brain adapts to ketones -> spares muscle protein (reduces gluconeogenesis demand)
SECTION 7: HORMONAL INTEGRATION
| Hormone | Effect on Carbohydrate | Effect on Lipid | Effect on Protein |
|---|---|---|---|
| Insulin | Increases glucose uptake, glycolysis, glycogen synthesis; inhibits gluconeogenesis | Inhibits lipolysis; stimulates FA synthesis, VLDL secretion | Anabolic; stimulates protein synthesis |
| Glucagon | Inhibits glycolysis; stimulates glycogenolysis, gluconeogenesis | Stimulates lipolysis (adipocytes); stimulates ketogenesis | Stimulates gluconeogenesis from AA |
| Cortisol | Stimulates gluconeogenesis | Stimulates lipolysis (permissive) | Catabolic; promotes protein breakdown -> AA for gluconeogenesis |
| Epinephrine | Stimulates glycogenolysis (muscle + liver); increases glycolysis | Stimulates lipolysis | Minor direct effect |
| Growth Hormone | Anti-insulin (decreases glucose uptake) | Stimulates lipolysis | Anabolic (IGF-1 mediated) |
| Thyroid hormone | Stimulates glycolysis, glycogenolysis | Stimulates lipolysis + oxidation | Catabolic in excess |
SECTION 8: CLINICAL CORRELATIONS (High-yield for NEET PG/USMLE/INICET)
1. Diabetic Ketoacidosis (DKA)
- Pathophysiology: Absolute insulin deficiency (Type 1 DM) -> glucagon unopposed
- Lipolysis maximal -> FFAs flood liver
- Gluconeogenesis maximal -> OAA depleted into PEP
- Acetyl-CoA accumulates (no OAA for TCA) -> ketone body formation
- Result: hyperglycemia + ketonemia + metabolic acidosis (HAGMA)
- Signs: Kussmaul respirations (compensatory respiratory alkalosis to blow off CO2), acetone breath, osmotic diuresis, dehydration
- Lab: Urine nitroprusside test detects acetoacetate (NOT beta-hydroxybutyrate - the predominant ketone in blood); low bicarbonate; elevated anion gap
- Tx: Insulin + IV fluids + potassium correction
2. Starvation Ketosis vs. DKA
| Feature | Starvation Ketosis | DKA |
|---|---|---|
| Blood glucose | Low/normal | Very high (>250 mg/dL) |
| Ketones | Mild-moderate | Severe |
| Insulin | Low but present | Near zero |
| pH | Mildly decreased | Severely decreased |
| Cause | Fasting/low carb diet | Uncontrolled T1DM |
3. Von Gierke Disease (Type Ia Glycogen Storage Disease)
- Defect: Glucose-6-phosphatase (liver, kidney, intestine)
- Cannot release free glucose from G6P -> cannot complete gluconeogenesis or glycogenolysis
- Features: Severe fasting hypoglycemia, hepatomegaly, lactic acidosis (lactate cannot be cleared), hyperuricemia (Cori cycle backs up -> increased lactate -> competes with urate for renal excretion), hyperlipidemia
4. McArdle Disease (Type V GSD)
- Defect: Muscle phosphorylase
- Cannot use muscle glycogen -> exercise intolerance, myoglobinuria
- Lactate does NOT rise with exercise (no lactate from muscle) -> used in forearm ischemic exercise test
5. Hyperammonemia / Urea Cycle Defects
- Ornithine transcarbamylase (OTC) deficiency: most common urea cycle defect (X-linked)
- Accumulation of ammonia -> neurological damage (ammonia inhibits alpha-KG -> blocks TCA -> CNS energy failure)
- Treatment: low-protein diet, nitrogen scavengers (sodium benzoate binds glycine; sodium phenylacetate binds glutamine)
6. Phenylketonuria (PKU)
- Defect: Phenylalanine hydroxylase (converts Phe -> Tyr)
- Phe accumulates -> converted to phenylpyruvate, phenylacetate, phenyllactate
- Blocks aromatic amino acid metabolism; competes with large neutral amino acids for brain transport
- Mental retardation, mousy/musty odor, hypopigmentation (reduced tyrosine -> less melanin)
- Treatment: phenylalanine-restricted diet; BH4 supplementation for mild forms
7. Maple Syrup Urine Disease (MSUD)
- Defect: Branched-chain alpha-keto acid dehydrogenase
- Accumulation of leucine, isoleucine, valine (and their keto acids)
- Maple syrup odor in urine, CNS toxicity, neonatal encephalopathy
- Leucine is most toxic (ketogenic -> excess ketones)
8. Methylmalonic Acidemia
- Defect: Methylmalonyl-CoA mutase (requires adenosyl-B12/cobalamin as cofactor)
- Propionyl-CoA -> methylmalonyl-CoA cannot be converted to succinyl-CoA -> cannot enter TCA
- Features: metabolic acidosis, hyperammonemia, ketosis, hypoglycemia
- Odd-chain fatty acids, isoleucine, valine, methionine, threonine are all affected (all feed into propionyl-CoA)
9. Alcoholic Ketoacidosis
- Ethanol metabolism produces excess NADH (alcohol dehydrogenase + aldehyde dehydrogenase)
- High NADH ratio: shifts OAA -> malate, pyruvate -> lactate
- OAA depleted -> TCA slows -> Acetyl-CoA accumulates -> ketogenesis
- Glucose does NOT rise (unlike DKA) because gluconeogenesis is also impaired (pyruvate/OAA depleted)
- Features: ketosis + lactic acidosis + hypoglycemia (no hyperglycemia)
10. Statin-Induced Myopathy
- HMG-CoA reductase inhibited -> blocks cholesterol synthesis
- Also depletes CoQ10 (ubiquinone, part of mevalonate pathway) -> mitochondrial dysfunction in muscle -> myopathy/rhabdomyolysis
11. Obesity and Metabolic Syndrome
- Chronic excess caloric intake: excess Acetyl-CoA -> fatty acid synthesis -> triglyceride storage
- Insulin resistance: hyperinsulinemia, hyperglycemia, dyslipidemia (high TG, low HDL, high LDL)
- Ectopic fat deposition in liver -> NAFLD/NASH
SECTION 9: PENTOSE PHOSPHATE PATHWAY INTEGRATION
- G6P can go to pentose phosphate pathway (PPP) instead of glycolysis
- PPP generates:
- NADPH: for FA synthesis, glutathione reductase (RBC oxidative defense), cytochrome P450
- Ribose-5-phosphate: for nucleotide synthesis
- G6PD deficiency: most common enzyme deficiency worldwide (X-linked); RBCs cannot regenerate NADPH -> oxidative hemolysis triggered by drugs (primaquine, dapsone), infections, fava beans
- Clinical: Heinz bodies (denatured Hb) + bite cells on smear; normal between episodes
SECTION 10: SUMMARY TABLE - KEY REGULATORY ENZYMES
| Pathway | Rate-Limiting Enzyme | Activated by | Inhibited by |
|---|---|---|---|
| Glycolysis | Phosphofructokinase-1 (PFK-1) | AMP, F2,6-BP, insulin | ATP, citrate, glucagon |
| Gluconeogenesis | PEPCK, Fructose-1,6-bisphosphatase | Glucagon, cortisol, cAMP | AMP, F2,6-BP, insulin |
| Glycogen synthesis | Glycogen synthase | Glucose, insulin (PP2A) | Glucagon, epinephrine (PKA phosphorylation) |
| Glycogenolysis | Glycogen phosphorylase | AMP, glucagon, epinephrine | Glucose, insulin |
| Fatty acid synthesis | Acetyl-CoA carboxylase | Insulin, citrate | Glucagon, palmitoyl-CoA, epinephrine |
| Beta-oxidation | CPT-1 (rate-limiting transport step) | Glucagon, low malonyl-CoA | Malonyl-CoA (fed state signal) |
| Ketogenesis | HMG-CoA synthase (mitochondrial) | Low malonyl-CoA, glucagon | High insulin, malonyl-CoA |
| TCA cycle | Isocitrate dehydrogenase | ADP, Ca2+, NAD+ | NADH, ATP |
| Urea cycle | Carbamoyl phosphate synthetase I (CPS-I) | N-acetylglutamate | Absent N-acetylglutamate |
| Cholesterol synthesis | HMG-CoA reductase | Insulin | Glucagon, sterols, statins |
QUICK REVISION - EXAM MEMORY AIDS
Purely Ketogenic AA: "Leu Lys" (L + L)
Mixed Ketogenic + Glucogenic: "I Pheel TWiTTy" = Isoleucine, Phenylalanine, Threonine, Tryptophan, Tyrosine
All others: Glucogenic
Gluconeogenesis cannot use:
- Even-chain fatty acids (Acetyl-CoA has no net contribution)
- Leucine, Lysine (purely ketogenic)
- Ethanol carbon directly
Can contribute to gluconeogenesis:
- ALL amino acids EXCEPT Leu and Lys
- Lactate, Pyruvate, Alanine
- Glycerol (from fat hydrolysis)
- Propionate/Odd-chain fatty acids (via succinyl-CoA)
Fasting blood glucose maintenance sequence:
- Glycogenolysis (0-6 hrs)
- Gluconeogenesis from lactate/alanine (6-24 hrs)
- Gluconeogenesis from muscle protein + Ketogenesis (1-3 days)
- Ketone adaptation by brain (3+ days) -> protein sparing
The CANNOT rules (high-yield):
- Fat CANNOT make net glucose (even-chain FA) - no glyoxylate cycle in humans
- RBCs CANNOT do beta-oxidation (no mitochondria)
- Muscle CANNOT release free glucose (no glucose-6-phosphatase)
- Brain CANNOT use fatty acids directly (FFA don't cross BBB well; uses glucose and after adaptation, ketones)
- Kidney CAN do gluconeogenesis (has glucose-6-phosphatase) - particularly during acidosis (uses glutamine)
Sources: Medical Physiology (Boron & Boulpaep), Basic Medical Biochemistry (Lieberman), 6e; Harper's Illustrated Biochemistry, 32e; Schwartz's Principles of Surgery, 11e
Compile all the cycles in one pdf correlating each other
Reading File
Running Command
ls /home/daytona/workspace/ 2>/dev/null && echo "---" && python3 -c "from reportlab.lib.pagesizes import A4; print('reportlab ok')" && python3 -c "from reportlab.platypus import SimpleDocTemplate; print('platypus ok')"
Running Command
mkdir -p /home/daytona/workspace/integrated-metabolism
This is a shared conversation. Sign in to Orris to start your own chat.