Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have all the material needed for a comprehensive, well-illustrated answer. Here it is:
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
ARDS is a syndrome of acute, diffuse, inflammatory lung injury resulting in increased pulmonary vascular permeability, loss of aerated lung tissue, and severe hypoxemia. The Berlin Definition requires: bilateral opacities on imaging, PaO2/FiO2 ratio <300 mmHg, onset within 7 days of a known insult, and respiratory failure not fully explained by cardiac failure or fluid overload.
Phases of ARDS
The pathologic progression follows three overlapping stages:

Time course of ARDS - Harrison's Principles of Internal Medicine, 22e
| Phase | Timing | Key Features |
|---|---|---|
| Exudative | Days 0-7 | Alveolar edema, neutrophil infiltration, hyaline membrane formation (diffuse alveolar damage - DAD) |
| Proliferative | Days 7-21 | Hyaline membrane resorption, interstitial inflammation, early fibrotic changes, type II pneumocyte hyperplasia |
| Fibrotic | >21 days (subset) | Pulmonary fibrosis, bullae formation, obliteration of capillaries |
Core Pathophysiology: Breakdown of the Alveolar-Capillary Barrier
The fundamental lesion in ARDS is disruption of the alveolar-capillary membrane, allowing protein-rich exudate to flood the alveolar space. Both the pulmonary microvascular endothelium and alveolar epithelium are involved.
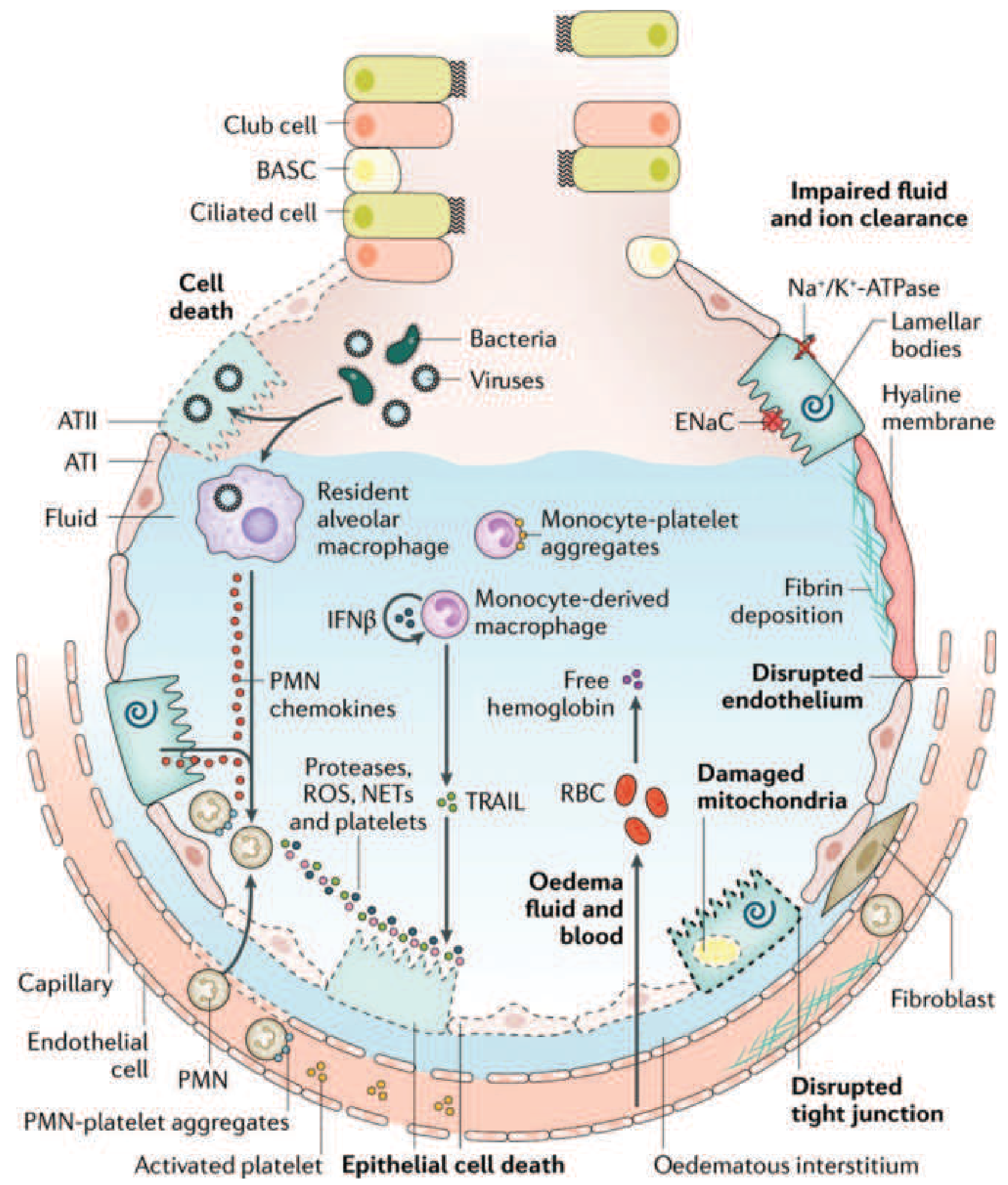
The injured alveolus in ARDS - Harrison's Principles of Internal Medicine, 22e (Fig. 312-3)
Step-by-Step Mechanistic Cascade
1. Initiating Insult: Direct vs. Indirect Lung Injury
ARDS arises from either pulmonary (direct) or extrapulmonary (indirect) triggers:
- Direct: pneumonia, aspiration of gastric contents, pulmonary contusion, toxic inhalation
- Indirect: sepsis (most common), major trauma, pancreatitis, massive transfusion (TRALI)
In indirect ARDS, circulating mediators (e.g., TNF-α, IL-1β, IL-8) reach the lung via the bloodstream and activate the local inflammatory cascade.
2. Innate Immune Activation
When pathogens or DAMPs (damage-associated molecular patterns) reach the alveolus, Toll-like receptors (TLRs) on alveolar macrophages and type I epithelial cells (ATI) are activated. This triggers nuclear factor-kB (NF-kB) signaling, driving production of:
- TNF-α and IL-1β - amplify the inflammatory response
- IL-8 (CXCL8) and MIP-2 - potent neutrophil chemoattractants
- Platelet-activating factor (PAF) - promotes neutrophil-endothelial adhesion
Alveolar macrophages also release IFN-β, which activates monocyte-derived macrophages recruited to the alveolus.
3. Neutrophil Sequestration and Transmigration
This is arguably the central effector mechanism:
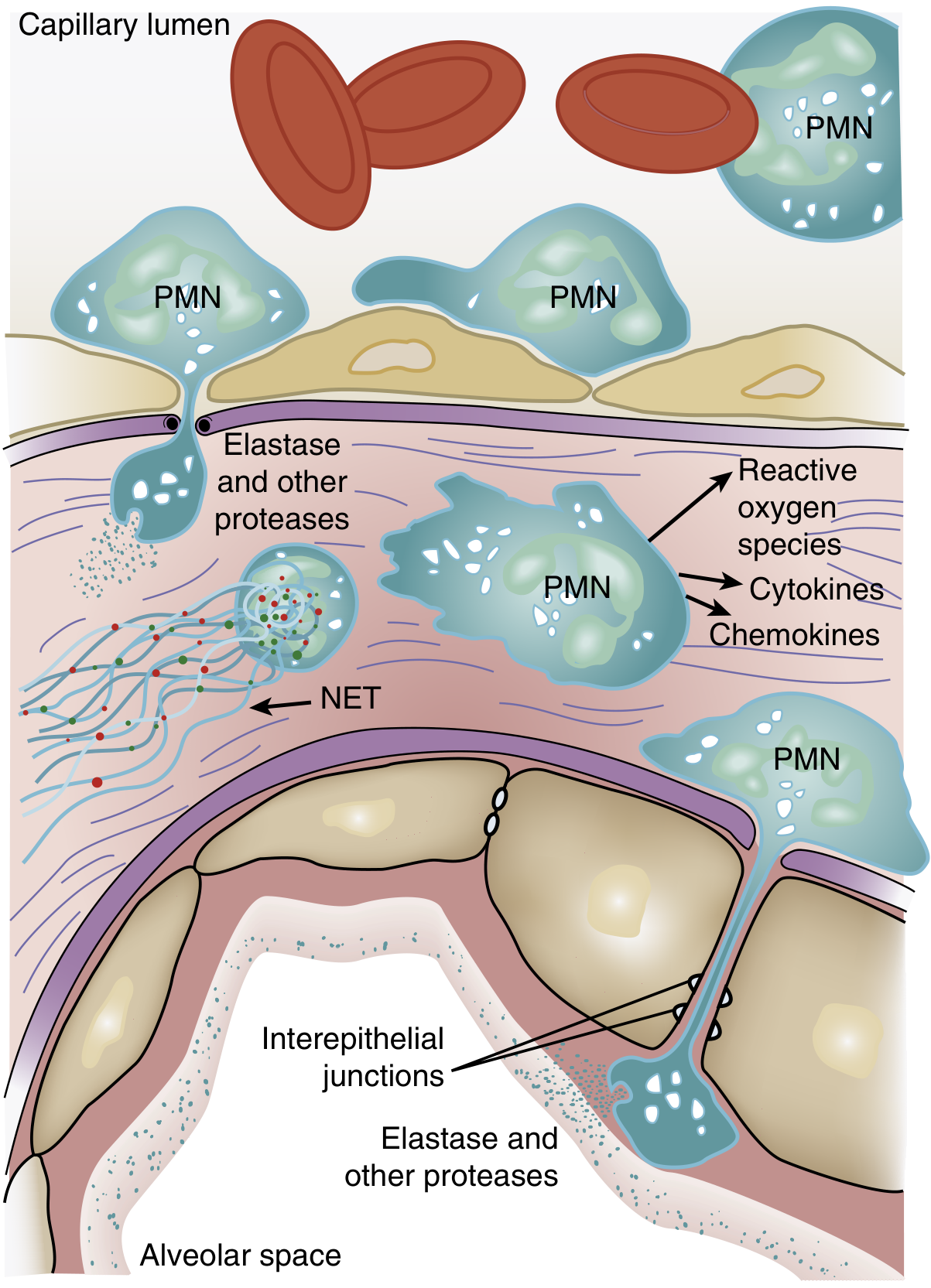
Role of neutrophils in ARDS - Murray & Nadel's Respiratory Medicine (Fig. 134.3)
- Pulmonary capillaries are narrower than the neutrophil diameter, so neutrophils must deform to pass through. Activated, "stiff" neutrophils (via actin cytoskeleton changes) cannot deform - they become sequestered in the microvasculature.
- One of the earliest signs of ARDS is a transient leukopenia from this pulmonary sequestration.
- Sequestered neutrophils then transmigrate across the endothelium into the interstitium and alveoli - this can occur without classical selectin/integrin-mediated adhesion once neutrophils are sequestered.
- Once in the alveolar space, activated neutrophils release a battery of injurious compounds:
| Compound | Mechanism of Injury |
|---|---|
| Reactive oxygen species (ROS) | Oxidative damage to lipid membranes, proteins, DNA |
| Neutrophil elastase (NE) | Degrades epithelial/endothelial cadherins (adherens junctions), destroying barrier integrity; cleaves cytokines and growth factors |
| Metalloproteinases | Matrix degradation, disruption of basement membrane |
| Defensins/cationic peptides | Direct cytotoxicity to epithelial/endothelial cells |
| TNF-α, IL-1β (released by neutrophils) | Further amplify the inflammatory cycle |
4. Neutrophil Extracellular Traps (NETs)
Activated neutrophils release NETs - web-like structures of DNA, histones, myeloperoxidase, NE, cathepsin G, and gelatinase. While designed to trap and kill pathogens, in ARDS, large-scale NET formation causes:
- Direct endothelial damage
- Promotion of thrombosis within pulmonary capillaries
- Amplification of the local cytokine storm
In animal LPS-instillation models of ARDS, treating with DNase (which degrades NETs) attenuates lung injury.
5. Alveolar Epithelial Injury
The type I pneumocytes (ATI) - which cover ~95% of the alveolar surface - are exquisitely sensitive to injury. Their damage disrupts the epithelial barrier and eliminates the tight junctions that normally exclude protein and fluid from the airspace. Consequences include:
- Alveolar flooding with protein-rich edema fluid
- Loss of surfactant function - phospholipase A2 (released from activated cells and pancreatic enzymes in pancreatitis) enzymatically degrades surfactant, promoting alveolar collapse and increased permeability
- Type II pneumocyte (ATII) dysfunction - normally responsible for surfactant synthesis and fluid clearance via Na⁺/K⁺-ATPase and ENaC (epithelial sodium channels). In ARDS, impaired ENaC and Na⁺/K⁺-ATPase activity leads to failure of alveolar fluid clearance, perpetuating the edema.
- ATII cells undergo death (apoptosis/necrosis), removing the progenitor pool for alveolar epithelial repair.
6. Endothelial Barrier Breakdown
Inflammatory mediators (TNF-α, thrombin, histamine, VEGF) cause:
- Loss of VE-cadherin and disruption of endothelial tight junctions
- Increased transcellular permeability via vesicle trafficking
- This allows protein-rich fluid, erythrocytes (free hemoglobin), and inflammatory cells to leak into the interstitium and alveolus
Free hemoglobin in the alveolus causes further oxidative damage.
7. Coagulation Dysregulation
The damaged alveolar-capillary interface activates the coagulation cascade:
- Exposure of subendothelial tissue factor activates the extrinsic pathway
- Fibrin deposition within the alveolus contributes to hyaline membrane formation - the pathologic hallmark of DAD
- Monocyte-platelet aggregates and activated platelets amplify local thrombosis and release more inflammatory mediators
- Intravascular fibrin can compress pulmonary capillaries, contributing to pulmonary hypertension
8. Pulmonary Hypertension
Multiple mechanisms combine to raise pulmonary artery pressure in ARDS:
- Hypoxic pulmonary vasoconstriction (HPV) in response to low alveolar PO2
- Intravascular fibrin deposition obstructing capillary flow
- Compression of blood vessels by positive-pressure mechanical ventilation
- Loss of capillaries from DAD in the fibrotic phase
9. Gas Exchange Failure
The net result of flooding and collapse:
- Right-to-left shunt: consolidated, fluid-filled alveoli receive perfusion but no ventilation - shunted blood returns deoxygenated to the systemic circulation
- Severe hypoxemia refractory to supplemental O2 (the defining feature of ARDS)
- Increased dead space: ventilated alveoli without adequate perfusion (due to microvascular injury) raise minute ventilation requirements and are measurable as elevated dead-space fraction (a predictor of mortality)
- Decreased compliance from flooded, collapsed, and stiff alveoli
Because the edematous lung behaves like a sponge, gravity redistributes fluid to dependent zones - creating three regions: consolidated/fluid-filled dependent zones, recruitable atelectatic zones, and a relatively spared non-dependent zone (the "baby lung" concept). This heterogeneity underlies ventilator-induced lung injury (VILI) risk.
10. Amplifying Loops and Genetic Susceptibility
- MAPK pathway: p38 MAPK activation by LPS stimulates TNF-α production and MIP-2 release, driving further neutrophil chemotaxis. Inhibiting p38 MAPK even hours after LPS exposure attenuates lung injury in mice.
- PI3-kinase-γ in neutrophils mediates their chemotaxis toward IL-8 and bacterial peptides.
- Renin-angiotensin system: ACE converts angiotensin I to angiotensin II (vasoconstrictor); ACE2 counteracts this. Loss of ACE2 (e.g., downregulation by SARS-CoV-1/2 via spike protein binding) tips the balance toward vasoconstriction and injury. This is a mechanistic basis for severe COVID-19-associated ARDS.
- Genetic risk factors: DARC gene (African Americans), NF-kB gene (white patients), PPFIA1 (trauma patients), ANG2 variants (risk via elevated angiopoietin-2 levels), and ACE insertion/deletion polymorphism.
Summary Diagram
The cascade can be summarized as:
Triggering insult (sepsis, aspiration, trauma, etc.)
↓
TLR activation → NF-kB → cytokine storm (TNF-α, IL-1β, IL-8)
↓
Neutrophil sequestration in pulmonary microvasculature
↓
Transmigration → ROS, elastase, NETs, proteases
↓
Alveolar-capillary barrier disruption
↓
Endothelial permeability ↑ + Epithelial injury (ATI/ATII)
↓
Protein-rich edema floods alveoli + Surfactant inactivated
↓
Coagulation activated → fibrin deposition → hyaline membranes
↓
Alveolar collapse, right-to-left shunt, dead-space ↑
↓
Refractory hypoxemia (PaO2/FiO2 <300) = ARDS
↓ (if unresolved)
Fibroproliferation → pulmonary fibrosis
Sources
- Murray & Nadel's Textbook of Respiratory Medicine, Chapter 134 (Pathology, Neutrophils, Alveolar-Capillary Membrane, Genetic Determinants)
- Harrison's Principles of Internal Medicine, 22e (2025), Chapter 312 (Clinical Course and Pathophysiology)
- Goldman-Cecil Medicine, Chapter 90/91 (ARDS, Lung-Protective Ventilation)
- Sabiston Textbook of Surgery (Berlin Definition, Management Principles)
This is a shared conversation. Sign in to Orris to start your own chat.