prepare about retinitis pigmentosa from basic to advances, clinical trials and recent advances in chat
"retinitis pigmentosa"[MeSH Terms] AND treatment
"retinitis pigmentosa" AND "gene therapy" AND clinical trial
retinitis pigmentosa recent advances clinical trials 2024 2025 gene therapy
"retinitis pigmentosa" AND optogenetics OR "stem cell" OR CRISPR
Retinitis Pigmentosa: From Basics to Advanced Clinical Knowledge
1. Definition and Overview
- Prevalence: 1 in 3,000 to 1 in 5,000 persons (approximately 1.5 million people globally)
- Age of onset: Variable - typically manifests in the first to third decade of life
- The severity at any given age varies significantly even among patients with the same gene mutation
2. Genetics and Molecular Basis
| Inheritance | Frequency | Key Genes | Prognosis |
|---|---|---|---|
| Autosomal Dominant (AD) | ~30-40% | RHO (rhodopsin), PRPF31, PRPH2 | Best prognosis |
| Autosomal Recessive (AR) | ~50-60% | USH2A, EYS, PDE6A, PDE6B, CNGB1 | Intermediate |
| X-linked (XL) | ~5-15% | RPGR (90% of XLRP), RP2 | Most severe |
| Digenic, mitochondrial | Rare | PRPH2 + ROM1 | Variable |
- Phototransduction cascade - rhodopsin (RHO) mutations are the most common cause of AD-RP; over 100 RHO mutations identified
- Retinoid cycle - RPE65, LRAT mutations
- Photoreceptor structure - PRPH2 (peripherin), ROM1
- Pre-mRNA splicing factors - PRPF3, PRPF8, PRPF31 (unique to retina despite ubiquitous expression)
- Ciliary transport - RPGR, RP2 (X-linked forms); the RPGR protein localizes to the connecting cilium of rod photoreceptors
3. Pathology and Pathophysiology
- Rod photoreceptors degenerate first → outer nuclear layer thinning
- Progressive RPE dysfunction → lipofuscin accumulation, phagocytosis failure
- Retinal pigment migration → pigment from RPE migrates perivascularly into the neural retina, forming "bone spicules"
- Retinal arteriolar attenuation → secondary to reduced metabolic demand from lost photoreceptors
- Optic nerve head atrophy → waxy pallor
- Secondary cone degeneration → loss of central acuity occurs later
4. Clinical Features
Symptoms (in typical order of onset)
- Nyctalopia (night blindness) - often the presenting symptom, due to rod loss
- Ring scotoma / peripheral visual field loss - mid-peripheral visual field defect that gradually expands concentrically
- Tunnel vision - characteristic "gun barrel" vision in advanced disease
- Photopsia - flickering or flashing lights
- Reduced central vision - a later feature (unless cataract intervenes earlier)
Classic Fundus Signs (the triad)
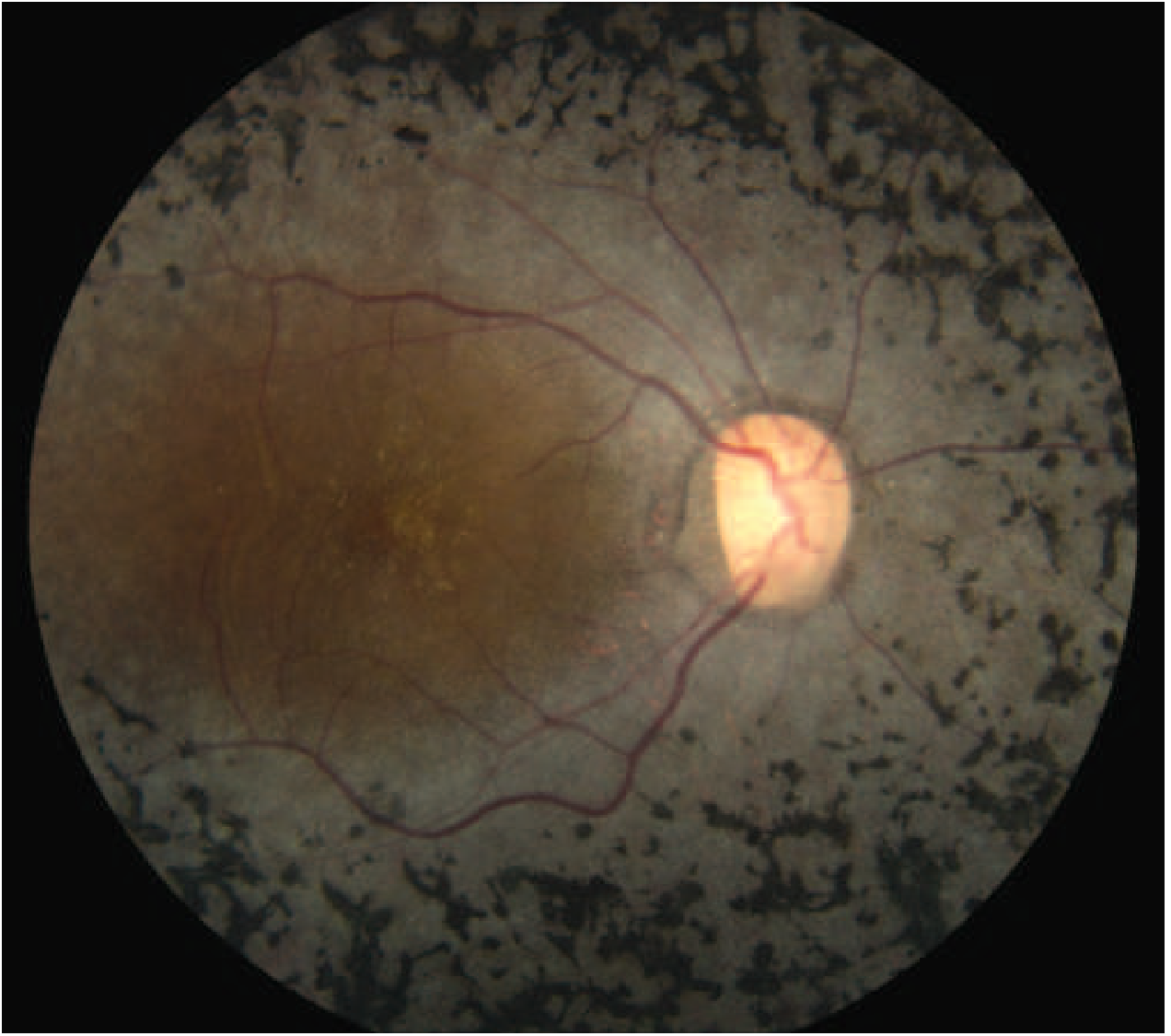
- Bone-spicule pigmentation - bilateral, mid-peripheral, intraretinal, perivascular. Named for resemblance to cancellous bone trabeculae. Starts sparse and expands anteriorly and posteriorly over time.
- Arteriolar attenuation - diffuse narrowing of retinal arterioles
- Waxy disc pallor - pale/yellowish optic disc (distinct from ischemic pallor)
Associated Ocular Features
- Posterior subcapsular cataract - common in all forms
- Cystoid macular oedema (CMO) - ~15% of patients; treatable cause of visual decline
- Epiretinal membrane (ERM) - contributes to metamorphopsia
- Macular atrophy - ~40% of patients on OCT
- Open-angle glaucoma - ~3%
- Optic disc drusen - increased frequency vs. general population
- Posterior vitreous detachment
Variant Forms
| Variant | Feature |
|---|---|
| RP sine pigmento | No bone spicules (early disease; all RP starts this way) |
| Sector RP | Pigmentation confined to one quadrant (usually inferior) |
| Unilateral RP | One eye involved; rule out inflammatory/toxic causes |
| Pericentral RP | Mid-peripheral scotoma sparing far periphery |
| XLRP female carrier | Golden "tapetal" macular reflex; centrifugal hyperautofluorescent lines on FAF |
5. Syndromic RP (20-30% of cases)
| Syndrome | System | Key Features |
|---|---|---|
| Usher syndrome | Hearing + retina | Most common cause of deaf-blindness; USH1, USH2, USH3 subtypes |
| Bardet-Biedl syndrome | Multi-system | Obesity, polydactyly, renal anomalies, cognitive impairment |
| Refsum disease | Metabolic | Phytanic acid accumulation; peripheral neuropathy, ataxia, ichthyosis |
| Kearns-Sayre syndrome | Mitochondrial | External ophthalmoplegia, cardiac conduction defects, onset <20 yrs |
| Bassen-Kornzweig (ABL) | Metabolic | Abetalipoproteinemia; fat malabsorption, spinocerebellar degeneration |
| NARP | Mitochondrial | Neuropathy, Ataxia, Retinitis Pigmentosa (mitochondrial point mutations) |
| Leber Congenital Amaurosis (LCA) | Retina-dominant | Severe early-onset variant (~5% of RP); RPE65, CEP290, GUCY2D mutations |
6. Differential Diagnosis / RP Mimics
- Drug toxicity: Chloroquine, hydroxychloroquine (bull's-eye maculopathy), thioridazine, chlorpromazine
- Infectious: Congenital rubella retinopathy, syphilitic chorioretinitis, CMV retinitis sequelae
- Traumatic: Old laser injury, retained intraocular foreign body
- Inflammatory: Diffuse unilateral subacute neuroretinitis (DUSN)
- Investigation for syphilis is sometimes warranted in atypical presentations
7. Investigations and Diagnosis
| Test | Findings |
|---|---|
| Full-field ERG | Diagnostic gold standard; extinguished or severely reduced scotopic (rod) response; photopic responses reduce with progression. Rarely needed in advanced disease. |
| Dark adaptation testing | Prolonged; useful in early/equivocal cases |
| Visual field (perimetry) | Mid-peripheral ring scotoma → concentric constriction → tunnel vision → central island → extinction |
| Microperimetry | Central visual function mapping; key endpoint in clinical trials |
| FAF (Fundus Autofluorescence) | Hyperautofluorescent perimacular ring (increased RPE lipofuscin); mid-peripheral hypo-AF patches. Distinguishes RP from normal fundus in ~95% of cases. |
| OCT | Quantifies macular involvement, ERM, CMO, and outer nuclear layer thinning; prognostic value |
| Genetic panel testing | Identifies mutation; facilitates genetic counseling and clinical trial eligibility |
8. Prognosis
- AD-RP: Best prognosis - many retain useful vision into middle age
- AR-RP: Intermediate severity
- XLRP: Most severe - central vision often reduced to 6/60 or less by the fifth decade
- Visual field loss is progressive and irreversible; rate varies widely by genotype
- CMO and ERM are treatable causes of visual decline within RP - identifying them is clinically important
9. Current Management
Approved Treatments
- Luxturna (voretigene neparvovec, AAV2-RPE65) - the first approved retinal gene therapy; FDA approved 2017 for biallelic RPE65 mutation-associated retinal dystrophy (covers LCA2 and RPE65-RP). Delivers a functional RPE65 gene subretinally. This is currently the only approved gene therapy for an RP subtype.
Supportive/Preventive Management
- Annual follow-up to detect treatable complications (CMO, ERM, cataract)
- Cataract surgery - often significantly beneficial
- CMO treatment - carbonic anhydrase inhibitors (acetazolamide, dorzolamide); anti-VEGF in refractory cases
- Low vision aids and rehabilitation services
- Smoking cessation - smoking worsens progression
- Vitamin A palmitate 15,000 IU daily - may slow rod ERG decline modestly (Goldman-Cecil); remains controversial; contraindicated in pregnancy; requires liver function monitoring
- Vitamin E should be avoided - may accelerate cone ERG decline
- DHA (docosahexaenoic acid) supplementation has not shown additional benefit in RCTs
- Genetic counseling for family members and reproductive planning
Retinal Prosthetics (Bionic Eye)
- Argus II (Second Sight) - epiretinal prosthesis; FDA approved 2013 for advanced RP; however the company went bankrupt in 2022, leaving patients without device support - a cautionary tale in device medicine
- Alpha AMS/IMS (Retina Implant AG) - subretinal photovoltaic prosthesis; European experience
10. Recent Advances and Active Clinical Trials (2023-2026)
A. Gene-Specific AAV Gene Therapy (Subretinal Delivery)
| Drug | Company | Trial | Status (2025-2026) |
|---|---|---|---|
| Botaretigene sparoparvovec (AAV5-RPGR, "bota-vec") | MeiraGTx / J&J | LUMEOS Phase 3 | Failed Phase 3 primary endpoint (May 2025) - a major setback |
| Cotoretigene toliparvovec (BIIB112, AAV8-RPGR) | Nightstar/Biogen | XIRIUS Phase 2/3 | Primary microperimetry endpoint not met (COVID impacted enrollment); significant LLVA improvement in low-dose arm [PMID: 38423215] |
| Laruparetigene zovaparvovec (laru-zova) | Beacon Therapeutics | SKYLINE/DAWN Phase 2/3 | Positive interim data - avg 16-letter LLVA improvement in 2nd eyes dosed in DAWN trial; pivotal Phase 2/3 results expected 2026 |
| AGTC-501 | Applied Genetic Technologies | HORIZON Phase 1/2 | 24-month positive safety and efficacy results published 2025 [PMID: 39643074] |
- AAV-PDE6A (Tübingen group) - Phase 1/2 subretinal gene supplementation; published safety and vision outcomes data 2026 [PMID: 40825661]
B. Gene-Agnostic / Modifier Gene Therapy
-
OCU400 (Ocugen) - AAV-NR2E3 (nuclear hormone receptor gene); acts as a "modifier" to regulate multiple retinal processes (photoreceptor development, metabolism, phototransduction, inflammation, survival). Phase 3 liMeliGhT trial fully enrolled with ~140 participants (70 RHO-RP + 70 other-gene RP); results expected early 2027. FDA clearance received 2024.
-
SPVN20 (SparingVision) - gene-agnostic therapy targeting cones; NYRVANA Phase 1/2 initiated in Belgium 2025-2026, expanding to France and Ireland.
C. Optogenetic Gene Therapy
-
MCO-010 / Visulyzr (Nanoscope Therapeutics) - multichannel opsin delivered to retinal cells; randomized controlled Phase 2/3 trial showed durable visual acuity gains maintained at 3 years. Rolling BLA submission to FDA initiated 2025; full submission planned H1 2026. Fast-track + orphan designation. First FDA approval of optogenetics may be imminent.
-
GS030 (GenSight Biologics) - ChrimsonR opsin + wearable goggle light stimulation device (PIONEER trial); Phase 1/2b ongoing follow-up.
-
ZM-02 (Zhongmou) - novel optogenetic approach; presented at ATC 2025 showing patients transitioning from complete blindness to functional vision (navigation capability, returning to cycling independently) in late-stage RP.
D. Antisense Oligonucleotides (ASO) and RNA Therapies
- Targeting dominant-negative RHO mutations with splice-switching ASOs
- QR-1123 (ProQR) - intravitreal ASO for P23H RHO-RP (most common AD-RP mutation in N. America); Phase 2/3 ILLUMINATE trial ongoing
- RNA base editing approaches being developed for dominant RHO variants
E. CRISPR / Gene Editing
- CRISPR-Cas9 editing for P23H rhodopsin (dominant negative) - approaches include "knock-and-replace" (silence mutant allele + deliver corrected copy)
- Subretinal CRISPR delivery proof-of-concept established in animal models
- No completed clinical trials yet in RP specifically, though LCA10 (CEP290) CRISPR editing (EDIT-101) established clinical feasibility in related disease
F. Cell-Based Therapies
-
Stem cell / retinal progenitor cells (jCell, jCyte Inc.) - intravitreal injection of human retinal progenitor cells; Phase 2 results showed visual improvement in some patients; neuroprotective rather than replacement mechanism proposed
-
iPSC-derived photoreceptor replacement (OpCT-001, BlueRock Therapeutics) - CLARICO trial: first-in-human iPSC-derived photoreceptor cell therapy; Phase 1 safety initiated July 2025 (up to 54 adults; dose-escalation design). A landmark in regenerative ophthalmology.
G. Neuroprotection
- CNTF (ciliary neurotrophic factor) - encapsulated cell technology (ECT) implants releasing CNTF; Phase 2 trials showed limited functional benefit despite some structural preservation
- Rod-derived cone viability factor (RdCVF) - TLQP-21 analog; neuroprotective for cones; being evaluated
- A 2026 systematic review (PMID: 42297463) examined neuroprotective therapies for visual acuity preservation - evidence remains limited for most agents
11. Key Trials Summary Table
| Trial | Drug | Mutation | Phase | Key Result |
|---|---|---|---|---|
| LUMEOS | Bota-vec (AAV5-RPGR) | XLRP | 3 | Failed primary endpoint (May 2025) |
| XIRIUS | Cotoretigene toliparvovec (AAV8-RPGR) | XLRP | 2/3 | LLVA improved (low dose); primary not met [38423215] |
| SKYLINE/DAWN | Laru-zova | XLRP | 2/3 | +16 letters LLVA; pivotal results expected 2026 |
| HORIZON | AGTC-501 | XLRP | 1/2 | 24-mo positive safety + efficacy [39643074] |
| liMeliGhT | OCU400 (AAV-NR2E3) | Multiple | 3 | Enrolled; results 2027 |
| PIONEER | GS030 | Any (advanced) | 1/2b | Ongoing |
| MCO-010 BLA | Visulyzr | Any (advanced) | Rolling BLA | FDA submission H1 2026 |
| ILLUMINATE | QR-1123 (ASO) | P23H RHO | 2/3 | Ongoing |
| CLARICO | OpCT-001 (iPSC) | Any (advanced) | 1 | First patient treated July 2025 |
| NYRVANA | SPVN20 | Any | 1/2 | Initiated 2025 |
| liMeliGhT | OCU400 | RHO + others | 3 | Results expected 2027 |
12. The Horizon: What's Coming
- Gene-agnostic therapies (optogenetics, modifier genes) are likely to reach patients before gene-specific approaches for most RP subtypes, simply because of scale and reduced need for genotyping
- MCO-010 (Nanoscope) is the closest to FDA approval for any RP treatment since Luxturna (2017) - and it targets all forms of late-stage RP
- iPSC-derived photoreceptor transplantation represents the theoretical endpoint - structural restoration of lost cells - but faces enormous challenges in synaptic integration
- Natural history studies and biomarker development (ellipsoid zone area on OCT, FAF ring diameter, mfERG) are refining clinical trial endpoints and patient selection
- Artificial intelligence is being applied to predict progression from retinal imaging and genotype-phenotype correlations
13. Quick Reference - High-Yield Points
- RP = rod-cone dystrophy, NOT inflammatory
- Classic triad: bone spicules + arteriolar attenuation + waxy disc pallor
- Night blindness → ring scotoma → tunnel vision → central loss
-
100 causative genes; RHO (AD), RPGR (XL), USH2A (AR) are most common
- Only approved RP-type gene therapy: Luxturna (RPE65 mutations)
- Treatable complications: CMO (CAI), cataract, ERM
- Vitamin A 15,000 IU/day may slow progression; avoid Vitamin E
- XLRP gene therapy trials active with bota-vec failure and laru-zova advancing
- Optogenetic therapy (MCO-010) in rolling FDA BLA - mutation-agnostic
- First iPSC photoreceptor cell trial (CLARICO) began July 2025
- Harrison's Principles of Internal Medicine 22E (2025), p. 275
- Kanski's Clinical Ophthalmology 10th Ed., p. 646-649
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 1912-1916
- Goldman-Cecil Medicine International Ed.
- Lam BL et al. XIRIUS trial. Ophthalmology 2024 [PMID: 38423215]
- Michaelides M et al. AAV5-RPGR Phase 1/2. Am J Ophthalmol 2024 [PMID: 38871269]
- Sherratt-Mayhew S et al. Neuroprotective therapies systematic review. BMJ Open Ophthalmol 2026 [PMID: 42297463]
- Yang P et al. AGTC-501 HORIZON 24-mo. Am J Ophthalmol 2025 [PMID: 39643074]
- Reichel FF et al. PDE6A subretinal gene therapy. Br J Ophthalmol 2026 [PMID: 40825661]
- Foundation Fighting Blindness / Drug Discovery News / Eye Health Institute 2025-2026
Create a visual comparison chart of active RP clinical trials (drugs, mechanisms, phases)
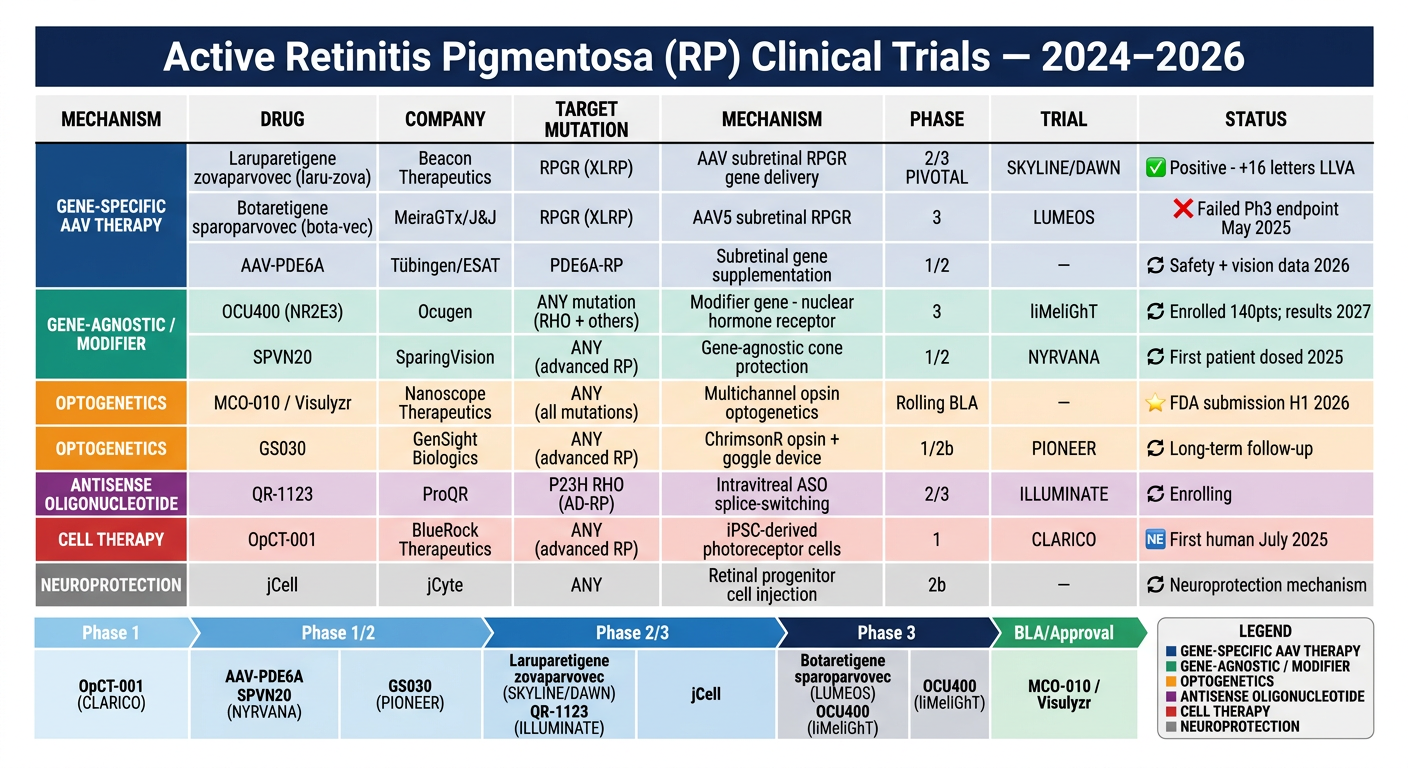
- MCO-010 (Visulyzr) by Nanoscope is furthest along overall - rolling BLA filed with FDA, full submission expected H1 2026. It's mutation-agnostic, meaning it could treat any RP patient with surviving inner retinal cells.
- Laru-zova (Beacon) is the strongest XLRP-specific candidate after bota-vec (MeiraGTx/J&J) failed its Phase 3 LUMEOS trial in May 2025 - a major setback for the field.
- OCU400 (Ocugen) is the only gene-agnostic modifier gene therapy in Phase 3, with 140 patients enrolled across RHO and non-RHO RP.
- CLARICO (BlueRock) - the world's first iPSC-derived photoreceptor cell transplant trial, with the first patient treated in July 2025.
in more details
retinitis pigmentosa clinical trials 2025 2026 MCO-010 OCU400 laru-zova CLARICO detailed results
"retinitis pigmentosa" AND "clinical trial" AND gene therapy
retinitis pigmentosa clinical trials details mechanism phase results 2025 2026
"retinitis pigmentosa" AND optogenetics AND MCO-010

Section 1 - Gene-Specific AAV Therapy (Mutation-Dependent)
Laru-zova (Laruparetigene zovaparvovec) - Beacon Therapeutics
- SKYLINE (Ph2): 36-month data presented EURETINA 2025 - durable microperimetry improvements in the high-dose arm, sustained through 3 years
- DAWN (Ph2): 15 men who had their first eye treated in SKYLINE received their second eye. Average gain: +16 ETDRS letters (3 lines) of low-luminance visual acuity (LLVA), plus microperimetry improvements
- VISTA (Ph2/3 PIVOTAL): Enrollment complete as of 2025; topline results expected H2 2026. Primary endpoint is LLVA, which is more sensitive than standard BCVA in dim conditions - the real-world relevant measure for XLRP patients
- LANDSCAPE trial: Newly launched study evaluating bilateral dosing safety (both eyes treated from the start)
Botaretigene sparoparvovec (bota-vec) - MeiraGTx
Cotoretigene toliparvovec (BIIB112) - Biogen
AAV-PDE6A - University of Tübingen
Section 2 - Gene-Agnostic / Modifier Therapy
OCU400 (NR2E3) - Ocugen
SPVN20 - SparingVision
Section 3 - Optogenetic Gene Therapy
MCO-010 / Visulyzr - Nanoscope Therapeutics
- Phase 1/2a (5-year follow-up, completed 2025): Safe and well tolerated throughout. No serious ocular adverse events.
- RESTORE Phase 2/3 RCT: Randomized, controlled, multicenter. Primary endpoint at Year 1: 40% of treated patients improved ≥10 ETDRS letters vs 0% sham (severely visually impaired patients - hand motions, light perception only at baseline).
- REMAIN extension (152 weeks = ~3 years): Average BCVA gain from baseline of +0.3 LogMAR (~15 letters, 3 lines) maintained at Week 152. BCVA area-under-the-curve was 5× greater than sham. Only 1 mild inflammation case requiring topical steroids; 14 of 15 treated patients needed no ongoing inflammation management.
- Presented at AAO 2025, Retina Society 2025, EURETINA 2025, Macula Society 2026 (Wills Eye Hospital's Dr. Allen Ho is a key investigator)
- FDA designations: Fast Track, Orphan Drug (RP + Stargardt disease)
- Rolling BLA initiated June 2025 - the first sections submitted; full package expected H1 2026
- Pipeline expansion: STARLIGHT Phase 2 in Stargardt disease complete; Phase 3 planned; preclinical programs in LCA and geographic atrophy
GS030 - GenSight Biologics
Section 4 - Antisense Oligonucleotide / RNA Therapy
QR-1123 - ProQR Therapeutics
Ultevursen - Sepul Bio
Section 5 - Cell-Based Therapy
OpCT-001 - BlueRock Therapeutics (Bayer)
jCell (Famzeretcel) - jCyte
DSP-3077 (Retinal Sheets) - Sumitomo Pharma
Section 6 - Neuroprotection & Small Molecules
N-Acetylcysteine (NAC) - Nacuity
RHO Small Molecule - Octant Bio
Bottom Line: Where the Field Stands in 2026
| Likelihood of approval | Drug | Why |
|---|---|---|
| Highest | MCO-010 (Nanoscope) | Rolling BLA filed; 3-yr RCT data solid; no genetic test needed |
| High (if 2026 data positive) | Laru-zova (Beacon) | VISTA results H2 2026; best XLRP candidate after bota-vec failure |
| 2027 target | OCU400 (Ocugen) | Phase 3 enrolled; data Q1 2027; gene-agnostic advantage |
| Early but promising | OpCT-001 (BlueRock) | First iPSC cell therapy; safety readout 2026-2027 |
| Niche but valuable | QR-1123 (ProQR) | Large P23H population; mutation-specific |
write in detail about retinitis pigmentosa
"retinitis pigmentosa" AND pathophysiology AND genetics
retinitis pigmentosa comprehensive review 2025 pathogenesis management epidemiology
"retinitis pigmentosa" AND management AND neuroprotection AND visual rehabilitation
"retinitis pigmentosa" AND "natural history" AND prognosis
Generate a comprehensive PDF study guide summarizing RP pathophysiology, genetics, and management