Old and new biomarkers in Heart failure with significance
Reading File
Finding Sources
Searching PubMed
"heart failure" AND "biomarkers" AND diagnosis prognosis
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive content from multiple authoritative textbooks. Let me compile the full, detailed answer.
Biomarkers in Heart Failure: Old and New
Heart failure (HF) biomarkers span the pathophysiological spectrum of the syndrome - from hemodynamic stress and myocyte injury to inflammation, fibrosis, and neurohormonal activation. A structured understanding is essential for diagnosis, prognosis, and monitoring.
I. Natriuretic Peptides (The Cornerstone Biomarkers)
BNP (B-type Natriuretic Peptide)
Biochemistry:
- BNP derives from a 134-amino acid precursor, preproBNP, rapidly cleaved to the 108-aa proBNP, and then by endoproteases furin and corin to a 32-aa C-terminal active fragment (BNP) and an inactive 76-aa N-terminal fragment (NT-proBNP), secreted in equimolar amounts.
- Secretion is driven by ventricular wall stretch and volume overload.
- BNP half-life: ~22 minutes (cleared by NPR-C receptor and endopeptidases).
- NT-proBNP half-life: 60-120 minutes (primarily renal clearance).
Physiological role: BNP has diuretic, natriuretic, and antihypertensive effects, and may protect against the fibrosis/remodeling that occurs in progressive HF.
Clinical cutoffs (Braunwald's, Fuster's, Tietz):
| Application | BNP | NT-proBNP |
|---|---|---|
| Exclude acute decompensated HF | <30-50 pg/mL (sens 97%, NPV 96%) | <300 pg/mL (sens 99%, NPV 99%) |
| Single cutoff to diagnose HF | 100 pg/mL (sens 90%, spec 76%, PPV 79%, NPV 89%) | - |
| Grey zone (uncertain) | 100-400 pg/mL | - |
| Rule in HF | >400 pg/mL (spec 91%) | - |
| Age-stratified NT-proBNP | - | <450 (age <50); <900 (age 50-75); <1800 (age >75) |
Breathing Not Properly Study: BNP at 100 pg/mL cutoff achieved sensitivity 90%, specificity 76%, PPV 79%, NPV 89% for HF in emergency dyspnea presentation.
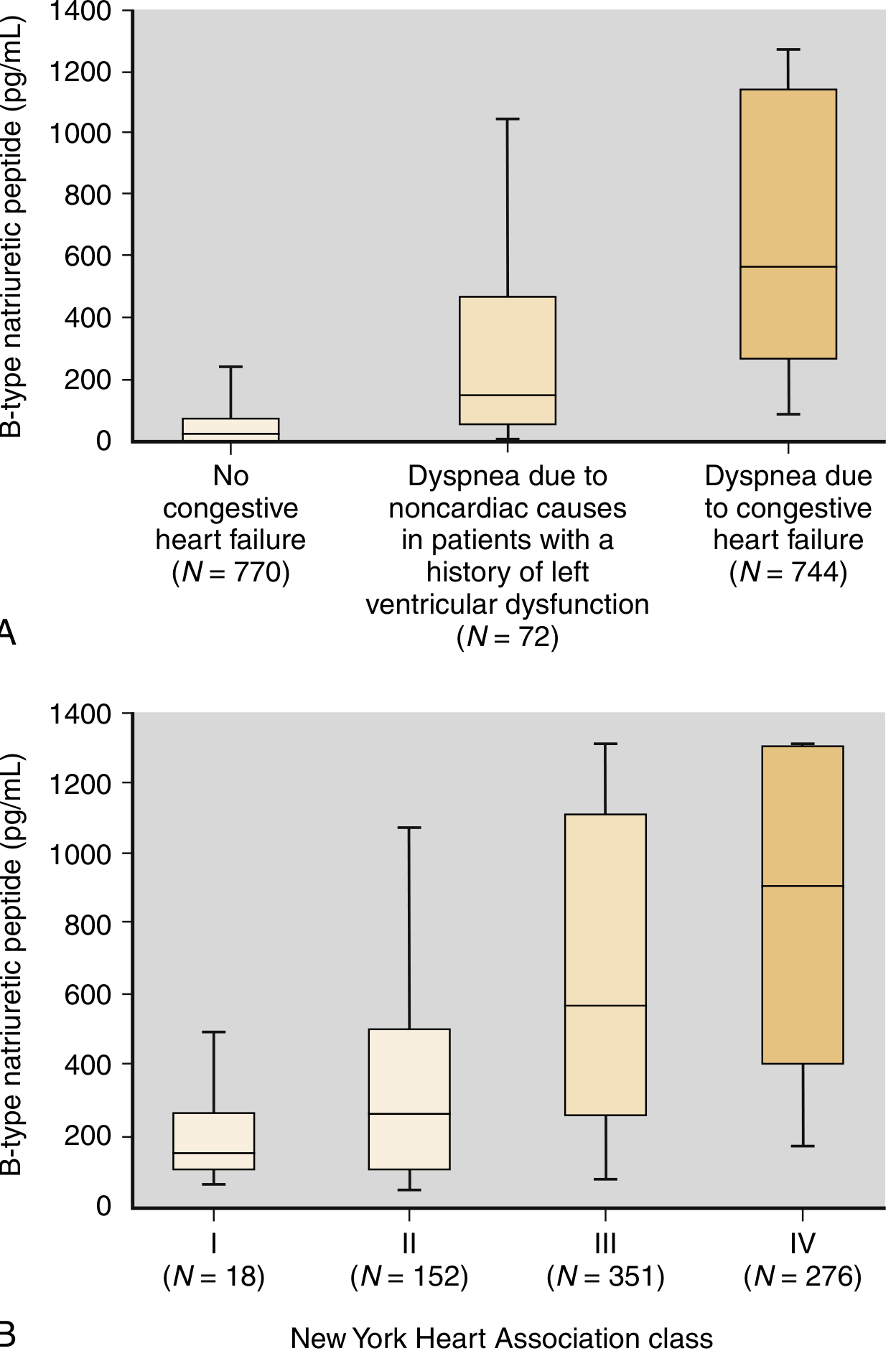
BNP levels rise progressively with NYHA class severity (I to IV) and clearly separate HF from non-cardiac dyspnea. - Henry's Clinical Diagnosis and Management by Laboratory Methods
ACC/AHA recommendation: Class I for diagnosis of acute HF and prognosis in chronic HF.
Serial testing: BNP/NT-proBNP decrease with effective treatment; serial testing combined with other markers (e.g., troponin) may assess treatment response and prognosis, though evidence for improved outcomes from serial BNP-guided therapy remains limited.
Important caveats (causes of elevated BNP):
- Cardiac: ACS, valve disease, other cardiomyopathies
- Non-cardiac: advancing age, anemia, renal failure, pulmonary disease, sleep apnea, critical illness, sepsis, severe burns
- Obesity and chronic stable HF can give falsely LOW levels (intraindividual variability 30-40%)
- Neprilysin inhibition (sacubitril/valsartan): raises BNP but NOT NT-proBNP; prefer NT-proBNP monitoring in these patients
NT-proBNP
- Higher circulating concentrations than BNP due to longer half-life
- Preferred marker when neprilysin inhibitors are used
- NT-proBNP <300 pg/mL makes acute HF diagnosis unlikely
- Age-stratified cutoffs improve diagnostic accuracy
MR-proANP (Mid-regional pro-Atrial Natriuretic Peptide)
- New test with some advantages over BNP/NT-proBNP
- Cutoff <57 pmol/L: sensitivity 98%, NPV 97% for excluding acute decompensated HF
- The rapidly responding atrial peptide and the slower-responding B-type NP may work synergistically - Tietz Textbook of Laboratory Medicine, 7th Edition, p.1822
II. Cardiac Troponins (Myocyte Injury Markers)
Context: Troponin elevation in HF indicates more severe disease. The role has evolved from purely ACS diagnosis to HF prognostication.
- Elevated cardiac troponin (cTn) in HF is associated with worse prognosis regardless of the degree of elevation: "more troponin elevation is always worse than less troponin elevation" - Tintinalli's Emergency Medicine
- High-sensitivity cardiac troponin (hs-cTnI/T) detects subclinical ongoing myocyte injury from necrosis, apoptosis, or reversible injury with increased membrane permeability
- In HF patients, elevated hs-cTn identifies those at greatest risk of adverse outcomes
- Current ACC/AHA recommendation: Class IIb - reasonable to measure in hospitalized HF patients to rule out ischemic trigger and establish prognosis
- Management implications of troponin elevations in chronic HF remain unclear
III. Soluble ST2 (sST2) - Fibrosis and Remodeling Marker
Mechanism:
- ST2 is a member of the Toll-like/interleukin-1 receptor superfamily with three isoforms.
- The transmembrane form: when bound by IL-33, produces antihypertensive and antifibrotic effects (cardioprotective).
- The soluble form (sST2): released in response to cardiomyocyte and macrophage stretch + inflammation. sST2 acts as a "decoy receptor" - binds circulating IL-33 and prevents it from binding to the transmembrane receptor, thus abolishing cardioprotection and promoting progressive fibrosis and remodeling.
- Strongly linked to progressive HF and death across all four ACC/AHA stages of HF.
Analytical details (Tietz):
- FDA-approved assay with LOD 1.3 ng/mL, LOQ 2.4 ng/mL
- Cutoff: 35 ng/mL (FDA; sex-specific values not required, though biologically warranted)
- Modest biological variation - advantage over BNP
Clinical significance:
- Less valuable as a diagnostic marker than BNP
- More prognostic than BNP for death and HF exacerbations
- Integrates both inflammatory and stretch-related processes
- Often remains prognostic even after accounting for BNP and other markers
- Reduced by beta-blockers and mineralocorticoid antagonists (therapies that improve outcomes)
- ACC/AHA Class IIb for risk stratification in chronic HF alongside galectin-3 and cardiac troponins
In acute ischemic heart disease: levels peak 6-18 hours after symptom onset; upper quartile values independently double the risk for cardiovascular death and HF.
IV. Galectin-3 - Fibrosis Marker
Mechanism:
- beta-Galactoside-binding lectin produced by macrophages in response to cellular injury
- Macrophages release galectin-3 which stimulates myofibroblasts to synthesize collagen, promoting cardiac fibrosis (maladaptive remodeling)
- Present in diverse tissues; elevated in many diseases (lower specificity)
Clinical significance:
- Predictive of adverse outcomes (death, HF exacerbations) in chronic HF
- Additive prognostic value to natriuretic peptides
- However, in head-to-head comparisons with ST2, galectin-3 has been "knocked out" of prognostic models in several studies
- Promising marker, but more treatment-related and intervention data are needed before routine clinical adoption
- Inhibited by mineralocorticoid antagonists and modified citrus pectin - Tietz Textbook of Laboratory Medicine, 7th Edition
V. Emerging / Novel Biomarkers
These are classified by the pathophysiological pathway they reflect (from Goldman-Cecil and Tietz):
Inflammation Markers
| Biomarker | Significance |
|---|---|
| C-reactive protein (CRP/hsCRP) | Reflects systemic inflammation; predicts outcomes but low HF-specificity |
| TNF-alpha | Elevated in HF; contributes to myocardial depression and remodeling |
| Interleukins (IL-1, IL-6, IL-18) | IL-6 may be more prognostic than hsCRP in some studies |
| Myeloperoxidase (MPO) | Released by neutrophils; elevated in active vascular inflammation; analytical stability issues limit routine use |
Neurohormonal Activation
| Biomarker | Significance |
|---|---|
| Copeptin | Stable surrogate for vasopressin; released in response to reduced cardiac output and plasma osmolarity changes. Vasopressin contributes to hyponatremia, vasoconstriction, and adverse cardiac remodeling |
| Endothelin-1 | Produced by endothelium in response to angiotensin II, inflammation, and shear stress; causes vasoconstriction and adverse cardiac remodeling |
Renal / Cardiorenal Markers
| Biomarker | Significance |
|---|---|
| Cystatin C | More sensitive and specific than creatinine for GFR changes; reflects cardiorenal syndrome; produced at constant rate by all nucleated cells |
| NGAL (Neutrophil Gelatinase-Associated Lipocalin) | Small glycoprotein released during inflammation and injury; involved in cell survival and matrix degradation; early marker of acute kidney injury in HF |
| BUN | More directly related to severity of acute HF than creatinine (integrates renal function + neurohormonal activation) |
Extracellular Matrix Remodeling
| Biomarker | Significance |
|---|---|
| Matrix metalloproteinases (MMPs) | Drive extracellular matrix degradation; associated with adverse LV remodeling |
| Tissue inhibitors of MMPs (TIMPs) | Counter-regulatory; imbalance with MMPs indicates fibrotic remodeling |
| Collagen propeptides (PICP, PINP) | Markers of collagen synthesis; reflect active myocardial fibrosis |
Growth Factors and Tissue Stress
| Biomarker | Significance |
|---|---|
| GDF-15 (Growth Differentiation Factor-15) | Member of TGF-beta superfamily; expressed in myocytes in response to ischemia, stretch, inflammation, and neurohormonal activation. Regulates inflammatory pathways, apoptosis, and cell repair. Predicts CVD and mortality in primary prevention populations. Component of the ABC score (with hs-cTn, NPs, and GDF-15) for bleeding prediction in AF patients |
| Neuregulin-1 | Growth factor important in cardiac development and pathogenesis of chronic HF; promotes cardiomyocyte growth and regulates the stress response |
| MR-proADM (Mid-regional pro-Adrenomedullin) | Produced by cardiovascular tissues in response to hemodynamic stress; favorable vascular effects (positive inotropy, vasodilation) |
Metabolic Markers
| Biomarker | Significance |
|---|---|
| Adiponectin | Adipokine with cardioprotective effects; paradoxically elevated in HF ("adiponectin paradox") |
| Chromogranin | Released from neuroendocrine cells alongside catecholamines; reflects adrenergic activation |
| Osteoprotegerin | Cytokine involved in bone metabolism and vascular calcification; elevated in HF with prognostic significance |
VI. Multimarker Strategy
From the Tietz Textbook and Goldman-Cecil:
"A number of additional biomarkers characterize inflammation, myocyte injury, neurohormonal upregulation, and extracellular matrix turnover in patients with heart failure. In the future, strategies that combine multiple biomarkers into a risk stratification model may prove additive to clinical judgment."
The multimarker concept recognizes that:
- No single marker captures the full complexity of HF pathophysiology
- Different biomarkers reflect different mechanisms (injury vs. stress vs. fibrosis vs. inflammation)
- Combining markers (e.g., NP + hs-cTn + ST2 or Galectin-3) provides additive prognostic information
VII. Summary Table: Old vs. New Biomarkers
| Category | Established ("Old") | Emerging ("New") |
|---|---|---|
| Hemodynamic stress | BNP, NT-proBNP | MR-proANP, MR-proADM, Copeptin |
| Myocyte injury | CK-MB, standard troponin | hs-cTnI/T, Heart-type FABP |
| Fibrosis/remodeling | - | ST2, Galectin-3, MMPs/TIMPs, Collagen propeptides |
| Neurohormonal | BNP | Copeptin (vasopressin surrogate), Endothelin-1 |
| Inflammation | CRP | hsCRP, IL-6, TNF-alpha, MPO, GDF-15 |
| Cardiorenal | Creatinine, BUN | Cystatin C, NGAL |
| Growth/repair | - | Neuregulin-1, GDF-15 |
VIII. Practical Decision Framework (ACC/AHA)
| Scenario | Recommended Biomarker | Recommendation Class |
|---|---|---|
| Diagnosis of acute HF (ED/hospital) | BNP or NT-proBNP | Class I |
| Prognosis in chronic HF | BNP or NT-proBNP | Class I |
| Risk stratification (chronic HF) | Troponin, ST2, Galectin-3 | Class IIb |
| Monitoring treatment response | NT-proBNP (preferred if on sacubitril/valsartan) | Evolving evidence |
| Cardiorenal assessment | Cystatin C, NGAL, BUN | Supportive use |
Recent evidence note: A 2025 systematic review and meta-analysis (PMID 39373821) confirmed that BNP and NT-proBNP are significant prognostic biomarkers for adverse outcomes specifically in HFpEF patients, reinforcing their value beyond HFrEF.
Sources: Henry's Clinical Diagnosis and Management by Laboratory Methods, p.318 | Tietz Textbook of Laboratory Medicine (7th ed.), p.1821-1822 | Fuster & Hurst's The Heart (15th ed.), p.1514 | Goldman-Cecil Medicine | Braunwald's Heart Disease (2 Vol.)
Whats is systolic dysfunction what is daistolic dysfunction What is hfref hfpef explain with mechanism
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a comprehensive, image-rich answer. Here it is:
Systolic vs. Diastolic Dysfunction & HFrEF vs. HFpEF
1. Systolic Dysfunction
Definition: Impaired ability of the ventricle to contract and eject blood during systole, resulting in a reduced ejection fraction (EF <40-50%).
Core problem: The ventricle cannot generate sufficient force to empty properly.
Mechanism:
- Loss of functional cardiomyocytes (MI, myocarditis, toxins)
- Impaired contractile protein function (cardiomyopathy, alcohol)
- Chronic pressure/volume overload overwhelming compensatory mechanisms
- Result: reduced stroke volume → reduced cardiac output → impaired tissue perfusion (forward failure)
- Residual blood pools backward → elevated filling pressures → pulmonary congestion, edema (backward failure)
Key changes:
- LV dilates (eccentric hypertrophy) - volume-overload pattern
- Wall becomes thin relative to chamber size
- EF falls (normal 50-70% → <40% in HFrEF)
- Increased end-systolic volume, increased end-diastolic volume
"Heart failure can result from progressive deterioration of myocardial contractile function (systolic dysfunction) - reflected as a decrease in left ventricular ejection fraction, the percentage of blood volume ejected from the ventricle during systole." - Robbins & Cotran Pathologic Basis of Disease
2. Diastolic Dysfunction
Definition: Impaired ability of the ventricle to relax and fill during diastole, with preserved contractile function (EF ≥50%).
Core problem: The ventricle is too stiff to receive blood normally.
Mechanism:
- Increased ventricular wall stiffness (LVH from hypertension, fibrosis, amyloid deposition, concentric remodeling)
- Abnormal myocardial relaxation due to dysfunctional calcium cycling
- Subendocardial fibrosis from chronic ischemia
- Loss of myocytes from aging → replaced by rigid collagen
Consequences:
- High filling pressures needed to push blood into a stiff LV (preload sensitivity)
- Elevated left atrial pressure → elevated pulmonary capillary wedge pressure → dyspnea
- Narrow fluid window: slight volume overload causes severe dyspnea; diuresis causes hypotension
- Atrial fibrillation is very poorly tolerated because atrial kick contributes disproportionately to filling a non-compliant ventricle
"Myocardial relaxation is an energy-dependent process. Processes that interfere with myocardial energy metabolism, such as ischemia, compromise myocardial relaxation. These changes reduce ventricular compliance and increase filling pressures." - Goldman-Cecil Medicine
Important note: Although we speak of "systolic" and "diastolic" HF, the majority of patients have both components to varying degrees. It is now preferable to classify patients as HFrEF or HFpEF rather than "systolic" or "diastolic" HF. - Goldman-Cecil Medicine
3. HFrEF - Heart Failure with Reduced Ejection Fraction
| Feature | Detail |
|---|---|
| EF | ≤40% |
| Also called | Systolic HF |
| Dominant defect | Impaired contraction |
| Ventricular morphology | Eccentric hypertrophy (dilated, thin-walled) |
| Typical causes | MI, dilated cardiomyopathy, myocarditis, valvular regurgitation, chronic volume overload |
Mechanism of HFrEF (Detailed)
Step 1 - Index event triggers a fall in cardiac output:
- Abrupt: myocardial infarction (loss of cardiomyocytes)
- Gradual: pressure or volume overload, genetic cardiomyopathy
- Hereditary: sarcomere gene mutations, dystrophin defects
Step 2 - Baroreceptors sense "underfilling" and activate compensatory mechanisms:
Step 3 - Neurohormonal Activation (the central pathophysiological driver):
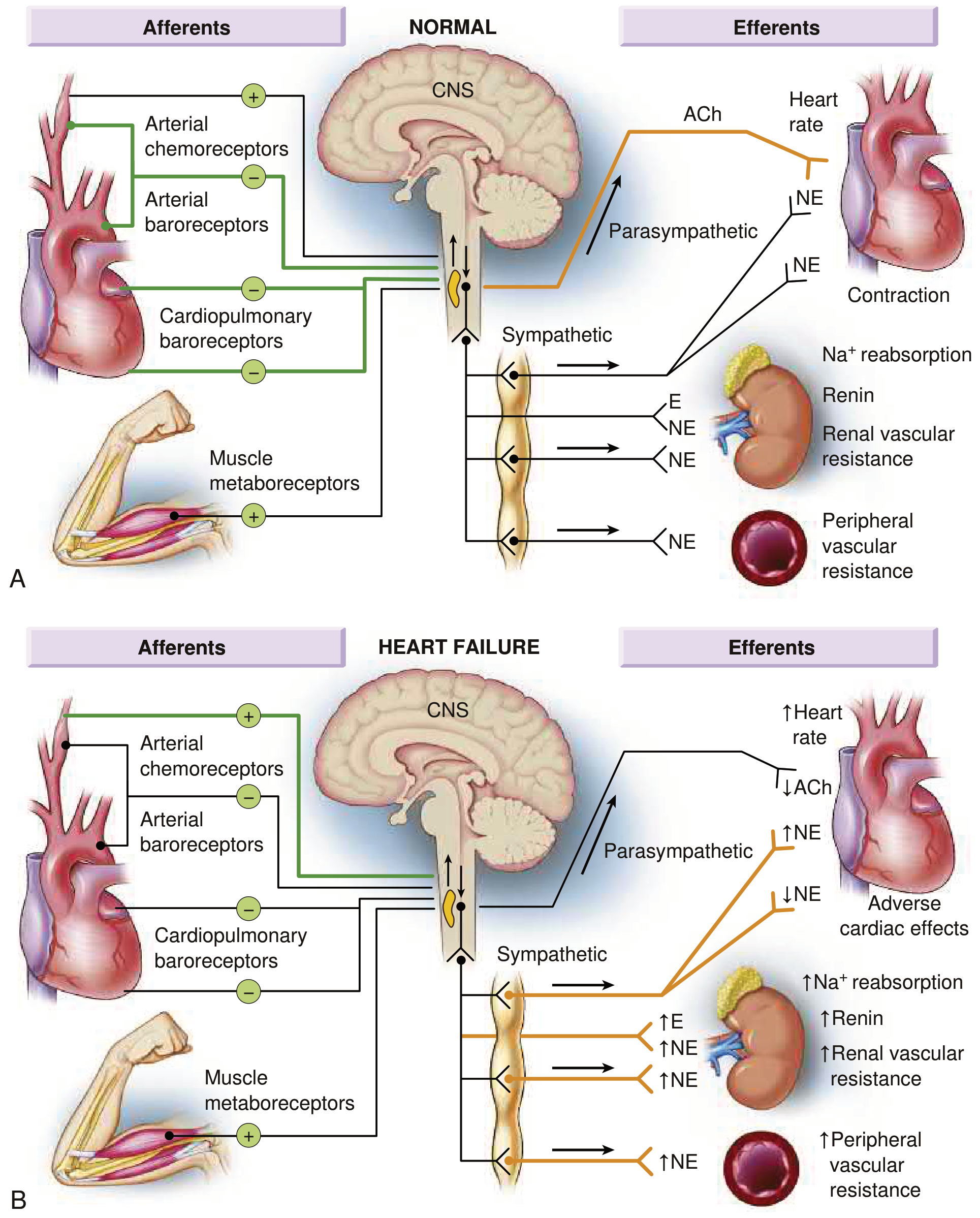
Normal (A) vs. Heart Failure (B): In HF, inhibitory input from baroreceptors and cardiopulmonary mechanoreceptors decreases while excitatory input increases, resulting in generalized sympathetic overactivation and parasympathetic withdrawal. - Braunwald's Heart Disease
A. Sympathetic Nervous System (SNS) Activation
- Baroreceptor inhibitory input decreases + excitatory chemoreceptor/metaboreceptor input increases
- Result: massive sympathetic outflow, parasympathetic withdrawal → loss of heart rate variability
- Circulating norepinephrine (NE) rises sharply
- Short-term benefit: increased HR, increased contractility, vasoconstriction → maintain BP
- Long-term harm: myocardial beta-1 receptor downregulation, direct myocyte toxicity, arrhythmias, further remodeling
B. RAAS Activation
- Reduced renal perfusion + sympathetic stimulation → renin release → angiotensin II (AngII) + aldosterone
- AngII: vasoconstriction, Na+ retention, stimulates NE release, promotes cardiac fibrosis
- Aldosterone: Na+ and water retention, promotes myocardial and vascular fibrosis
- Short-term benefit: volume retention increases preload, maintains CO
- Long-term harm: volume/pressure overload, fibrosis, progressive LV dysfunction
C. ADH (Vasopressin) / Copeptin
- Released in response to reduced CO and elevated AngII
- Promotes free water retention → hyponatremia (a poor prognostic sign)
- Contributes to vasoconstriction
Step 4 - LV Remodeling (the structural consequence of neurohormonal toxicity):
LV remodeling = collective molecular, cellular, and structural changes in response to injury and altered loading.
| Change | Mechanism | Consequence |
|---|---|---|
| Cardiomyocyte hypertrophy | Increased sarcomere assembly in series | LV dilation (eccentric) |
| Cardiomyocyte apoptosis | Oxidative stress, AngII, TNF-alpha | Loss of myocytes, thinning |
| Interstitial fibrosis | Aldosterone, AngII, TGF-beta | Increased stiffness, impaired relaxation |
| Beta-receptor downregulation | Chronic NE excess | Blunted inotropic reserve |
| Fetal gene re-expression | Myosin heavy chain switch (alpha → beta) | Reduced contractility |
| Mitochondrial dysfunction | Energy substrate shift | Impaired ATP generation |
The end result is a dilated, thin-walled, globally hypocontractile ventricle with markedly reduced EF - the hallmark of HFrEF.
Step 5 - Vicious cycle: Remodeling further reduces cardiac output → more neurohormonal activation → more remodeling (progressive deterioration unless interrupted by therapy targeting RAAS/SNS).
4. HFpEF - Heart Failure with Preserved Ejection Fraction
| Feature | Detail |
|---|---|
| EF | ≥50% |
| Also called | Diastolic HF |
| Dominant defect | Impaired relaxation and filling |
| Ventricular morphology | Concentric hypertrophy (thick-walled, small or normal cavity) |
| Typical patients | Older, female, obese, hypertensive, diabetic, with AF |
Mechanism of HFpEF (Detailed)
HFpEF was once viewed as "simply" diastolic dysfunction, but it is now recognized as a complex multifactorial systemic illness involving aging, inflammation, multimorbidity, and lifestyle factors. - Braunwald's Heart Disease
Core pathophysiological mechanisms:
1. Increased Ventricular Stiffness
Two types of stiffness contribute:
a) Passive (structural) stiffness:
- Hypertension → concentric LV hypertrophy → parallel sarcomere assembly → thick walls, small cavity
- Shifts in collagen isoforms (type I > type III) → more rigid cross-linking
- Myocardial fibrosis (driven by aldosterone, AngII, TGF-beta, galectin-3)
- Amyloid deposition, glycogen accumulation
b) Active (kinetic) stiffness - impaired relaxation:
- Relaxation is an energy-dependent process requiring active Ca²+ reuptake by SERCA2a into the SR
- In HFpEF: SERCA2a activity reduced, phospholamban inhibition increased → slow, incomplete Ca²+ removal → slow ventricular relaxation
- Ischemia impairs energy supply → worsens relaxation
- Titin isoform shift (N2B stiff isoform predominates) → increased myofibrillar resting tension
2. Elevated Filling Pressures
- Stiff ventricle requires higher filling pressure to achieve adequate end-diastolic volume
- Left atrial pressure rises → pulmonary venous hypertension → dyspnea
- The resting hemodynamic profile can be normal; physiologic stress (exercise, tachycardia) provokes exaggerated filling pressure elevation → exertional limitations
3. Systemic Inflammation and Microvascular Dysfunction
- Comorbidities (obesity, diabetes, hypertension, CKD) drive systemic inflammation
- Coronary microvascular inflammation → reduced NO bioavailability → impaired myocardial relaxation
- Endothelial dysfunction → impaired coronary vasodilation on demand
- This inflammatory paradigm distinguishes HFpEF from a "pure" diastolic problem
4. Chronotropic Incompetence and AF
- Atrial fibrillation is very poorly tolerated - atrial kick contributes disproportionately to filling a non-compliant ventricle
- Loss of atrial contraction worsens filling further
- Tachycardia shortens diastolic filling time → worsens symptoms acutely
5. Extracardiac Contributions
- Arterial stiffness (from aging and hypertension) increases pulsatile afterload
- Peripheral skeletal muscle dysfunction and abnormal oxygen extraction contribute to exercise intolerance
- Renal dysfunction limits diuretic responsiveness
- Obesity and metabolic syndrome amplify inflammation and pericardial restraint
5. Head-to-Head Comparison: HFrEF vs. HFpEF
| Feature | HFrEF | HFpEF |
|---|---|---|
| EF | <40% | ≥50% (HFmrEF: 41-49%) |
| Primary defect | Impaired contraction (systole) | Impaired relaxation/filling (diastole) |
| Ventricular morphology | Eccentric hypertrophy (dilated) | Concentric hypertrophy (thick walls) |
| Wall thickness | Normal or thin | Increased |
| Cavity size | Enlarged | Normal or small |
| Typical demographics | Younger, male, post-MI | Older, female, obese, hypertensive |
| Common causes | MI, dilated CMP, myocarditis | Hypertension, DM, obesity, aging, amyloidosis |
| Neurohormonal activation | Prominent (SNS, RAAS) | Present but less dominant |
| Response to disease-modifying therapy | ACEi/ARB/ARNI, beta-blockers, MRA, SGLT2i (all reduce mortality) | SGLT2i (reduce hospitalizations); others less proven |
| Prognosis | Poor (but improving with therapy) | Somewhat better but hospitalization similar; no proven mortality-reducing therapy except SGLT2i |
| Key echocardiographic findings | Reduced LVEF, global hypokinesis, dilated LV | Preserved LVEF, concentric LVH, impaired E/e', elevated filling pressures |
| BNP/NT-proBNP | Elevated | Also elevated (lower values in obese) |
"It is preferable to characterize patients as having heart failure with a preserved ejection fraction or heart failure with a reduced ejection fraction rather than as having systolic or diastolic heart failure." - Goldman-Cecil Medicine
6. Why They Share Symptoms Despite Different Mechanisms
Both HFrEF and HFpEF produce elevated filling pressures (though by different means) → elevated pulmonary venous pressure → pulmonary congestion → dyspnea, orthopnea, PND, edema.
- HFrEF: Filling pressures elevated because a failing dilated ventricle retains residual volume (backward failure)
- HFpEF: Filling pressures elevated because high pressure is needed to push blood into a stiff ventricle
The downstream clinical consequences (dyspnea, reduced exercise tolerance, hospitalization for fluid overload) are therefore similar in both conditions, which is why differentiation requires echocardiography and/or NP measurement.
Sources: Robbins & Cotran Pathologic Basis of Disease | Braunwald's Heart Disease (2 Vol.) | Goldman-Cecil Medicine | Tintinalli's Emergency Medicine | Tietz Textbook of Laboratory Medicine
Which drugs given in which class of heart failure
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
I now have all the information needed for a comprehensive, well-structured answer. Let me compile it.
Drugs in Heart Failure by Class
Heart failure therapy follows two parallel classification systems - the ACC/AHA Stages (A-D) and NYHA Functional Classes (I-IV). Understanding both is essential because drugs are prescribed based on both structural disease and symptom severity.
Classification Systems at a Glance
| ACC/AHA Stage | NYHA Class | Description | Goal |
|---|---|---|---|
| Stage A | Pre-failure | At risk, no structural disease, no symptoms | Prevention |
| Stage B | Class I | Structural disease, no symptoms | Prevent progression |
| Stage C | Class II/III | Structural disease + current/prior symptoms | Reduce mortality + symptoms |
| Stage D | Class IV | Refractory HF, symptoms at rest | Palliation / Advanced therapies |
"Stage C patients have structural heart disease and symptoms of failure, and symptoms are responsive to ordinary therapy... Stage D patients have heart failure refractory to ordinary therapy, and special interventions are required." - Katzung's Basic & Clinical Pharmacology
Drug Assignment by Stage
STAGE A - "At Risk" (Hypertension, DM, CAD, cardiotoxic drug exposure, family history of cardiomyopathy)
No structural disease, no symptoms. Goal: PREVENT heart failure from developing.
| Drug Class | Examples | Rationale |
|---|---|---|
| Antihypertensives | ACEi, ARBs, CCBs, thiazides | Treat HTN - the #1 modifiable risk factor |
| ACE Inhibitors | Enalapril, ramipril | Reduce risk in high-risk patients (HTN, DM) |
| SGLT2 Inhibitors | Dapagliflozin, empagliflozin | Reduce HF risk in DM patients |
| Statins | Atorvastatin, rosuvastatin | Treat dyslipidemia, reduce atherosclerosis |
| Lifestyle modification | - | Weight loss, exercise, smoking cessation, alcohol reduction, glycemic control |
STAGE B / NYHA CLASS I - "Pre-Heart Failure" (Structural disease, e.g., LVH, low EF, prior MI - but ASYMPTOMATIC)
Goal: Prevent transition to symptomatic HF and sudden death.
| Drug Class | Examples | Indication |
|---|---|---|
| ACE Inhibitor | Enalapril, lisinopril, ramipril | Reduce ventricular remodeling, slow progression; all patients with reduced LVEF |
| ARB (if ACEi intolerant) | Valsartan, candesartan | Same benefit as ACEi |
| Beta-Blocker | Bisoprolol, carvedilol, metoprolol CR | All patients with reduced LVEF after MI; slow remodeling |
| SGLT2 Inhibitor | Dapagliflozin, empagliflozin | Now indicated for asymptomatic LV dysfunction |
| ICD | Device | Patients with LVEF ≤35% post-MI at risk for sudden cardiac death |
STAGE C / NYHA CLASS II-III - Symptomatic HFrEF (LVEF ≤40%)
Goal: Reduce mortality, reduce hospitalizations, improve symptoms and quality of life.
This is where the "Four Pillars of HFrEF" come in - all four should be started simultaneously and up-titrated as quickly as tolerated:
"Four life-saving foundational therapies should be prescribed in any order for all patients, wherever possible: an angiotensin receptor neprilysin inhibitor, a β-blocker, a mineralocorticoid receptor antagonist, and an SGLT2 inhibitor." - Goldman-Cecil Medicine
PILLAR 1 - ARNI (preferred) or ACEi/ARB
| Drug | Starting Dose | Target Dose |
|---|---|---|
| Sacubitril/Valsartan (ARNI) | 24/26 mg BD | 97/103 mg BD |
| Enalapril (ACEi) | 2.5 mg BD | 10-20 mg BD |
| Ramipril (ACEi) | 1.25-2.5 mg OD | 10 mg OD |
| Candesartan (ARB) | 4-8 mg OD | 32 mg OD |
Mechanism: ACEi/ARBs block the RAAS - reduce preload, afterload, and cardiac remodeling. ARNI additionally blocks neprilysin (raises BNP, ANP) - superior to ACEi alone (PARADIGM-HF trial).
Note: Cannot combine ACEi + ARB + ARNI together. Switch ACEi/ARB to ARNI in NYHA II/III.
PILLAR 2 - Beta-Blockers
| Drug | Starting Dose | Target Dose |
|---|---|---|
| Bisoprolol | 1.25 mg OD | 10 mg OD |
| Carvedilol | 3.125 mg BD | 25-50 mg BD |
| Metoprolol CR/XL | 12.5-25 mg OD | 200 mg OD |
| Nebivolol | 1.25 mg OD | 10 mg OD |
Mechanism: Block the chronically over-activated SNS. Reduce HR, reduce myocardial oxygen demand, prevent arrhythmias, reverse pathological remodeling (the "reverse remodeling" effect).
CAUTION: Start ONLY in EUVOLEMIC, clinically stable patients. Do NOT start in acute decompensated HF or when signs of congestion are present. Contraindicated in asthma, 2nd/3rd degree AV block.
Rule: "Start low, go slow, aim high" - double dose at 2-week intervals minimum.
PILLAR 3 - Mineralocorticoid Receptor Antagonist (MRA)
| Drug | Starting Dose | Target Dose |
|---|---|---|
| Spironolactone | 25 mg OD or alternate days | 25-50 mg OD |
| Eplerenone | 25 mg OD | 50 mg OD |
Indication: NYHA class II-IV, LVEF ≤40%, added to ARNI/ACEi/ARB + beta-blocker + SGLT2i.
Mechanism: Block aldosterone - reduce Na+ retention, prevent myocardial and vascular fibrosis (reduces galectin-3, TGF-beta activity).
CAUTION: Check K+ and renal function at 1, 4, 8, 12 weeks then 6-monthly. Stop or reduce dose if K+ >5.5 mmol/L or creatinine >2.5 mg/dL.
PILLAR 4 - SGLT2 Inhibitors
| Drug | Starting Dose | Target Dose |
|---|---|---|
| Dapagliflozin | 10 mg OD | 10 mg OD |
| Empagliflozin | 10 mg OD | 10 mg OD |
Indication: NYHA class II-IV, LVEF ≤40%, first-line regardless of diabetes status.
Mechanism (multiple): Osmotic diuresis/natriuresis, reduce preload/afterload, reduce visceral adiposity, improve mitochondrial function, reduce inflammation, possible direct cardioprotective effects.
CONTRAINDICATION: eGFR <20 mL/min/1.73m², DKA history. Use caution if eGFR <30.
ADDITIONAL DRUGS (Stage C - Selected Patients)
| Drug | Indication/Class | Key Details |
|---|---|---|
| Diuretics (Loop) | All symptomatic patients with congestion | Furosemide, torasemide - relieves dyspnea/edema rapidly; does NOT reduce mortality but essential for symptom control |
| Thiazides | Mild congestion, preserved renal function | Chlorthalidone, hydrochlorothiazide - used if loop diuretics not yet needed |
| Ivabradine | NYHA II-III, SR, HR ≥70 bpm despite max beta-blocker | Blocks If current in SA node - purely rate-reducing, reduces HF hospitalizations (SHIFT trial) |
| Hydralazine + Isosorbide dinitrate | African American patients with NYHA III-IV despite optimal therapy | Fixed combination 37.5mg/20mg TDS → target 75mg/40mg TDS. FDA approved specifically in Black patients. Also used if ARNI/ACEi/ARB intolerant |
| Digoxin | Persistent symptoms NYHA II-IV, or AF with rapid ventricular response | Reduces hospitalizations, improves symptoms; no mortality benefit (DIG trial). Therapeutic range 0.5-1.0 ng/mL |
| Vericiguat | NYHA II-IV, recently worsened HF | sGC stimulator - increases cGMP, reduces vascular resistance; approved for patients recently hospitalized or IV diuretics given |
| Omega-3 PUFA | NYHA II-IV | 1g daily n-3 PUFA - modest reduction in mortality/hospitalizations |
| ICD (implantable) | LVEF ≤35%, NYHA II-III, on optimal therapy ≥3 months | Prevents sudden cardiac death |
| CRT | LVEF ≤35%, LBBB, QRS ≥150ms, NYHA II-IV | Cardiac resynchronization therapy - improves symptoms and mortality |
STAGE D / NYHA CLASS IV - Advanced / Refractory HF
Symptoms at rest, refractory to all standard medical therapy.
| Intervention | Details |
|---|---|
| Maximize all 4 pillars | Continue/optimize ARNI + BB + MRA + SGLT2i if tolerated |
| IV diuretics | High-dose furosemide IV, bumetanide, torsemide |
| IV inotropes (short-term) | Dobutamine (beta-1 agonist), milrinone (PDE3 inhibitor) - for cardiogenic shock/bridge to transplant; do NOT reduce mortality |
| Vasopressin antagonists | Tolvaptan - for hyponatremia in advanced HF |
| LVAD (Left Ventricular Assist Device) | Bridge to transplant OR destination therapy (continuous-flow devices); improves survival in end-stage HF |
| Heart Transplantation | Definitive therapy; highly selected patients, age usually <65-70 |
| Palliative/compassionate care | When above options exhausted or declined |
HFpEF (NYHA Class II-IV, EF ≥50%) - Drugs
| Drug | Evidence | Role |
|---|---|---|
| SGLT2i (dapagliflozin, empagliflozin) | Proven (EMPEROR-Preserved, DELIVER) | Reduce HF hospitalizations + improve QoL - only class with proven benefit |
| Diuretics | Symptom control only | Reduce congestion; use with caution (narrow fluid window) |
| Spironolactone | TOPCAT trial - modest benefit in Americas | Reduce HF hospitalizations (17% reduction); no clear mortality benefit |
| ARNi (sacubitril/valsartan) | PARAGON-HF - near-significant | Reduces symptoms; may benefit women and EF near lower range |
| Beta-blockers / Rate-limiting CCBs | If AF present | Control ventricular rate in AF - critical as atrial kick is important in stiff LV |
| ACEi/ARB | May help reduce LVH | Not proven to reduce mortality in isolated HFpEF |
| Treat underlying causes | HTN, DM, ischemia, AF | Most impactful intervention; BP control is the cornerstone |
Summary Drug-to-Class Table (HFrEF)
| Drug | Stage A | Stage B | Stage C (NYHA II-III) | Stage D (NYHA IV) |
|---|---|---|---|---|
| ACEi | ✓ | ✓ | ✓ | ✓ |
| ARB | - | ✓ | ✓ | ✓ |
| ARNI (sacubitril/valsartan) | - | - | ✓ (preferred over ACEi/ARB) | ✓ |
| Beta-blocker | - | ✓ (post-MI) | ✓ | ✓ |
| MRA | - | - | ✓ | ✓ |
| SGLT2i | ✓ (DM) | ✓ | ✓ | - |
| Loop diuretic | - | - | ✓ (if congested) | ✓ |
| Ivabradine | - | - | ✓ (HR ≥70, sinus rhythm) | ✓ |
| Hydralazine/nitrate | - | - | ✓ (African American, or ARNI/ACEi intolerant) | ✓ |
| Digoxin | - | - | ✓ (persistent symptoms/AF) | ✓ |
| Vericiguat | - | - | ✓ (recently worsened) | ✓ |
| IV inotropes | - | - | - | ✓ (short-term/bridge) |
| LVAD / Transplant | - | - | - | ✓ |
Key clinical rule from Goldman-Cecil: "Start low, go slow, aim high" - initiate all four pillars quickly at low doses, up-titrate to evidence-based target doses. Some beta-blocker is always better than none.
Sources: Goldman-Cecil Medicine, p.477-492 | Katzung's Basic & Clinical Pharmacology (16th ed.), p.344-345 | Fuster & Hurst's The Heart (15th ed.) | Braunwald's Heart Disease (2 Vol.)
This is a shared conversation. Sign in to Orris to start your own chat.