Pickwickian syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
obesity hypoventilation syndrome Pickwickian
Reading File
Searching PubMed
obesity hypoventilation syndrome treatment outcomes
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a thorough answer. Here is the full overview:
Pickwickian Syndrome (Obesity Hypoventilation Syndrome)
Origin of the Name
The term "Pickwickian syndrome" comes from the character Joe in Charles Dickens's The Posthumous Papers of the Pickwick Club (1837) - a fat, loud-snoring, perpetually sleepy boy. Sir William Osler in 1918 was the first to formally link obesity with this presentation. Burwell et al. in 1956 added the key association with alveolar hypoventilation, and Drachman & Gummit in 1962 described periodic cessation of respiration. Today the preferred clinical term is Obesity Hypoventilation Syndrome (OHS).
Definition
OHS is defined by three criteria (all must be present):
- Obesity (BMI ≥ 30 kg/m²)
- Chronic alveolar hypoventilation with daytime hypercapnia (awake PaCO₂ ≥ 45 mm Hg)
- Sleep-related breathing disorder
- Absence of other causes of hypoventilation (it is a diagnosis of exclusion)
Epidemiology
- Prevalence of 10-20% in obese OSA patients referred to sleep centers
- Nearly 50% in hospitalized patients with BMI ≥ 50 kg/m²
- Approximately 0.3% in the general U.S. adult population
- Prevalence is rising as super-obesity (BMI ≥ 50) increases
Pathophysiology
The development of hypercapnia in OHS results from a complex interplay of three mechanisms:
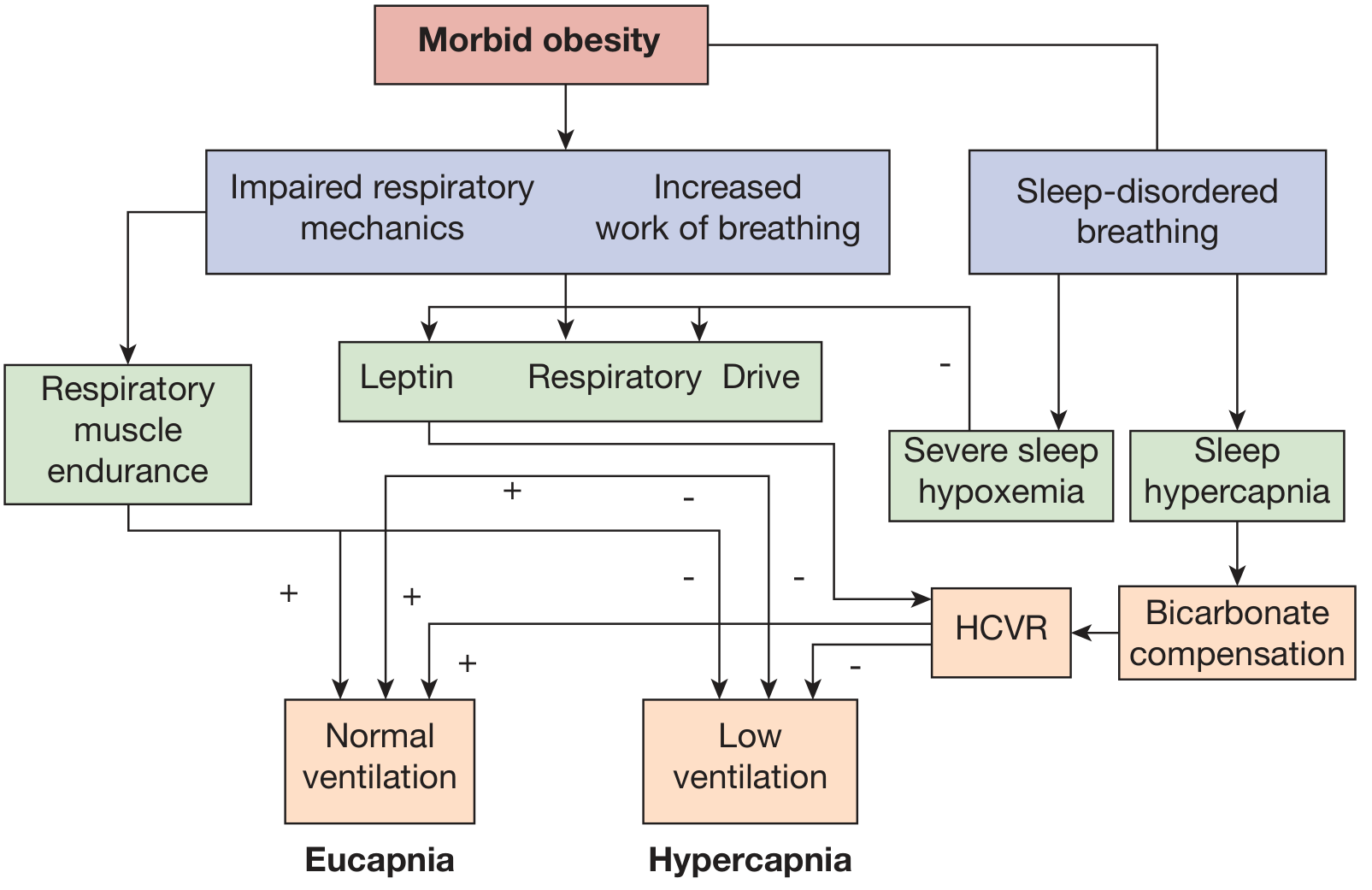
1. Abnormal respiratory mechanics / increased work of breathing:
- Excess abdominal and perithoracic fat displaces the diaphragm rostrally, reducing its mechanical efficiency
- Reduced chest wall and respiratory system compliance
- Increased airway resistance and expiratory flow limitation
- Intrinsic PEEP, basilar atelectasis, and V/Q mismatch
2. Sleep-related breathing disorder:
- ~90% of OHS patients have co-existing OSA
- During obstructive events, CO₂ loads accumulate; compensatory hyperventilation between events becomes progressively inadequate over time
- During REM sleep, muscle atonia further worsens hypoventilation
3. Blunted central ventilatory drive:
- Blunted hypercapnic ventilatory response (HCVR)
- Blunted hypoxic ventilatory response
- Central hypothalamic "resistance" to leptin (leptin is an adipokine that normally stimulates ventilation; leptin resistance impairs this protective effect)
- Renal bicarbonate retention as metabolic compensation for chronic CO₂ retention further blunts the central drive to breathe
Clinical Presentation
Symptoms:
- Excessive daytime sleepiness (hallmark) - even beyond what OSA alone explains
- Morning headaches (from nocturnal hypercapnia)
- Loud snoring, witnessed apneas
- Dyspnea on exertion (more prominent than in eucapnic OSA)
Signs on examination:
- Morbid obesity
- Facial plethora, injected sclerae
- Prominent pulmonic component of S2 (pulmonary hypertension)
- Lower extremity edema (right heart failure / cor pulmonale)
- Awake hypoxemia: PaO₂ typically < 70 mm Hg (uncommon in eucapnic OSA)
Associated morbidity compared to eucapnic obese patients:
- Higher rates of systemic hypertension, angina, congestive heart failure
- Cor pulmonale and pulmonary hypertension
- Polycythemia (secondary erythrocytosis from chronic hypoxia)
- Increased ICU admissions; can present in acute-on-chronic hypercapnic respiratory failure
Diagnosis
| Step | Finding |
|---|---|
| Arterial blood gas (ABG) | PaCO₂ ≥ 45 mm Hg while awake, on room air - essential for diagnosis |
| Serum bicarbonate | ≥ 27 mEq/L is highly sensitive (92%) but not specific (50%) for chronic hypercapnia; useful for screening |
| Pulse oximetry | SpO₂ < 94% in an obese patient with OSA should prompt ABG |
| Polysomnography (PSG) | Prolonged deep desaturations not typical of OSA pattern; SpO₂ < 90% for extended periods; > 10 mm Hg rise in PaCO₂ during sleep |
| Pulmonary function tests | Restrictive pattern; reduced ERV (expiratory reserve volume) |
| ECG | Signs of right heart strain / cor pulmonale |
| Thyroid function | To exclude hypothyroidism as a cause |
Causes of hypoventilation to exclude: significant pulmonary disease, chest wall disorders, neuromuscular disease, severe hypothyroidism, other central hypoventilation syndromes.
Treatment
1. Positive Airway Pressure (PAP) - First-line
CPAP:
- Effective in 60-80% of OHS patients (particularly those with moderate-to-severe OSA phenotype / high AHI)
- Improves hypercapnia by unloading accumulated CO₂ from repetitive obstructive events
- Improves sleep quality, daytime sleepiness, and quality of life
- If hypercapnia persists after 2 months of adherent CPAP, transition to NIPPV
NIPPV (BiPAP / bilevel PAP):
- Indicated for: low AHI phenotype, CPAP non-responders, acute decompensation
- Typical settings: EPAP 6-10 cm H₂O, IPAP 16-20 cm H₂O, pressure support difference ≥ 8-10 cm H₂O
- A backup respiratory rate (set ~2 breaths/min below waking RR) is recommended given ventilatory control abnormalities
- Prolonged inspiratory times are important to allow uniform tidal volume delivery and atelectasis recruitment
Volume Assured Pressure Support (VAPS):
- Automatically adjusts pressure support to maintain a target tidal volume (e.g. when effort decreases in REM sleep)
- Useful for OHS but data comparing it to standard BiPAP are limited
2. Oxygen Supplementation
- Required in up to 50% of OHS patients in addition to PAP to maintain SpO₂ > 90%
- Oxygen alone is contraindicated - it can prolong apnea duration and worsen hypercapnia
- Oxygen must always be combined with NIPPV
3. Weight Loss
- Should be encouraged in all patients; even modest weight loss improves outcomes
- Often difficult to sustain with lifestyle measures alone
- Newer agents such as semaglutide (GLP-1 agonist) are showing promise
- Bariatric surgery can lead to significant improvement or resolution of OHS and should be considered in eligible patients
Key Distinguishing Points vs. OSA Alone
| Feature | OSA alone | OHS |
|---|---|---|
| Daytime CO₂ | Normal | Elevated (≥ 45 mm Hg) |
| Awake SpO₂ | Normal | Often < 94% |
| Morning headaches | Uncommon | Common |
| Cor pulmonale | Rare | More common |
| Polycythemia | Rare | More common |
| Serum HCO₃⁻ | Normal | Often ≥ 27 mEq/L |
| CPAP response | Usually complete | 60-80%; may need BiPAP |
Prognosis
OHS carries significantly higher morbidity and mortality than eucapnic obesity or OSA alone. Timely diagnosis and adherent PAP therapy improve outcomes substantially. Untreated, it progresses to pulmonary hypertension, right heart failure, and recurrent acute hypercapnic respiratory failure.
Sources: Fishman's Pulmonary Diseases and Disorders, 2-Volume Set (Chapter 99); Miller's Anesthesia 10e; Bradley and Daroff's Neurology in Clinical Practice
Alports
Finding Sources
Searching PubMed
Alport syndrome
Reading File
Reading File
Reading Article
PMID: 39673454
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is the comprehensive overview:
Alport Syndrome
Definition
Alport syndrome (AS) is an inherited disorder of basement membranes caused by mutations in the type IV collagen genes (COL4A3, COL4A4, or COL4A5), resulting in a defective or absent α3α4α5(IV) collagen network. It is characterized by:
- Progressive nephritis → end-stage kidney disease (ESKD)
- Sensorineural hearing loss
- Ocular abnormalities
It is the second most common monogenic kidney disorder after polycystic kidney disease, affecting approximately 1 in 50,000 people, and accounts for ~0.5% of adults and 1.7% of children with ESKD in the United States.
Molecular Basis - Type IV Collagen
The GBM contains three type IV collagen chains assembled as a heterotrimer: α3(IV)-α4(IV)-α5(IV). This network is also present in Bowman's capsule, distal tubular BM, alveolar BM, the cochlea, and the eye lens.
- The six COL4A genes are arranged in back-to-back pairs on three chromosomes:
- COL4A1/COL4A2 → chromosome 13
- COL4A3/COL4A4 → chromosome 2
- COL4A5/COL4A6 → X chromosome (Xq22-24)
Mutations disrupt the proper folding of these chains into triple helices, destabilizing the entire network.
Genetics - Three Forms
| Form | Gene(s) | Inheritance | Frequency |
|---|---|---|---|
| X-linked (XLAS) | COL4A5 | X-linked | ~60-70% |
| Autosomal recessive (ARAS) | COL4A3 or COL4A4 | AR | ~15% |
| Autosomal dominant (ADAS) | COL4A3 or COL4A4 (heterozygous) | AD | ~20-30% (more common with NGS) |
XLAS in males: The most severe form. Risk of ESKD in untreated males:
- 50% by age 30
- 80% by age 40
- Nearly 100% by age 60
Genotype-phenotype correlation (males):
- Truncating variants (large deletions, nonsense mutations) → ESKD before age 30 in up to 90%
- Missense / splice variants → slower progression, ESKD often after age 30
XLAS in females: Wide variability due to random X-inactivation (lyonization). Most have only microhematuria, but up to 25% develop significant kidney manifestations. Hematuria is seen in 95%, proteinuria in 75%, hearing loss in 28%, ocular defects in 15%.
ARAS: Both sexes equally affected; most reach ESKD and deafness before age 30 regardless of sex.
Clinical Features
Kidney
- Persistent microscopic hematuria - the universal presenting feature (often from childhood)
- Episodic gross hematuria, often triggered by respiratory infections or exercise, with flank or abdominal pain
- Proteinuria - mild early, increases progressively; nephrotic syndrome can occur
- Hypertension - a late manifestation
- Progressive renal failure → ESKD, typically between ages 16-35 in X-linked males
Hearing
- High-frequency sensorineural deafness in 30-50% of patients
- The lesion is cochlear (shown by brainstem auditory-evoked responses)
- Hearing loss is always accompanied by renal involvement, but severity does not correlate with renal disease severity
- Some families have hereditary nephritis without deafness
Ocular (15-30%)
- Anterior lenticonus (protrusion of the central lens into the anterior capsule) - virtually pathognomonic of Alport syndrome
- "Dot and fleck" macular retinopathy
- Keratoconus, spherophakia, myopia, cataracts, retinitis pigmentosa
Other (rare)
- Aortic dissections, aneurysms, dilation, aortic insufficiency
- Diffuse esophageal/tracheobronchial leiomyomatosis (associated with large COL4A5-COL4A6 deletions)
- Epstein syndrome (megathrombocytopenia + nephritis)
- Fechtner syndrome (nephritis + macrothrombocytopenia + Döhle-like inclusions + deafness + cataract)
Pathology
Light Microscopy (non-specific)
- Early (<5 years): may be normal; occasional superficial fetal glomeruli or interstitial foam cells
- Later: mesangial proliferation, matrix increase, irregular GBM thickening and lamellation
- Advanced: focal segmental glomerulosclerosis (FSGS) pattern, tubular atrophy, interstitial fibrosis
- Note: a significant proportion of familial FSGS without apparent GBM lamellation may harbor COL4 mutations
Immunofluorescence (key diagnostic tool)
- Often negative (no immune complex deposits - helps exclude immune GN)
- Commercially available antisera to collagen IV α-chains show:
- In X-linked males: absent α3, α4, α5 from GBM; absent α5 from Bowman's capsule, distal TBM, and skin
- In XLAS females: segmental/mosaic loss of α5 in GBM and skin (due to X-inactivation)
- In ARAS: absent α3, α4, α5 from GBM, but α5 retained in Bowman's capsule and skin (because α5 pairs with α6 there)
→ Skin biopsy showing absence of α5(IV) is highly specific for X-linked AS and can diagnose some males non-invasively.
Electron Microscopy (definitive)
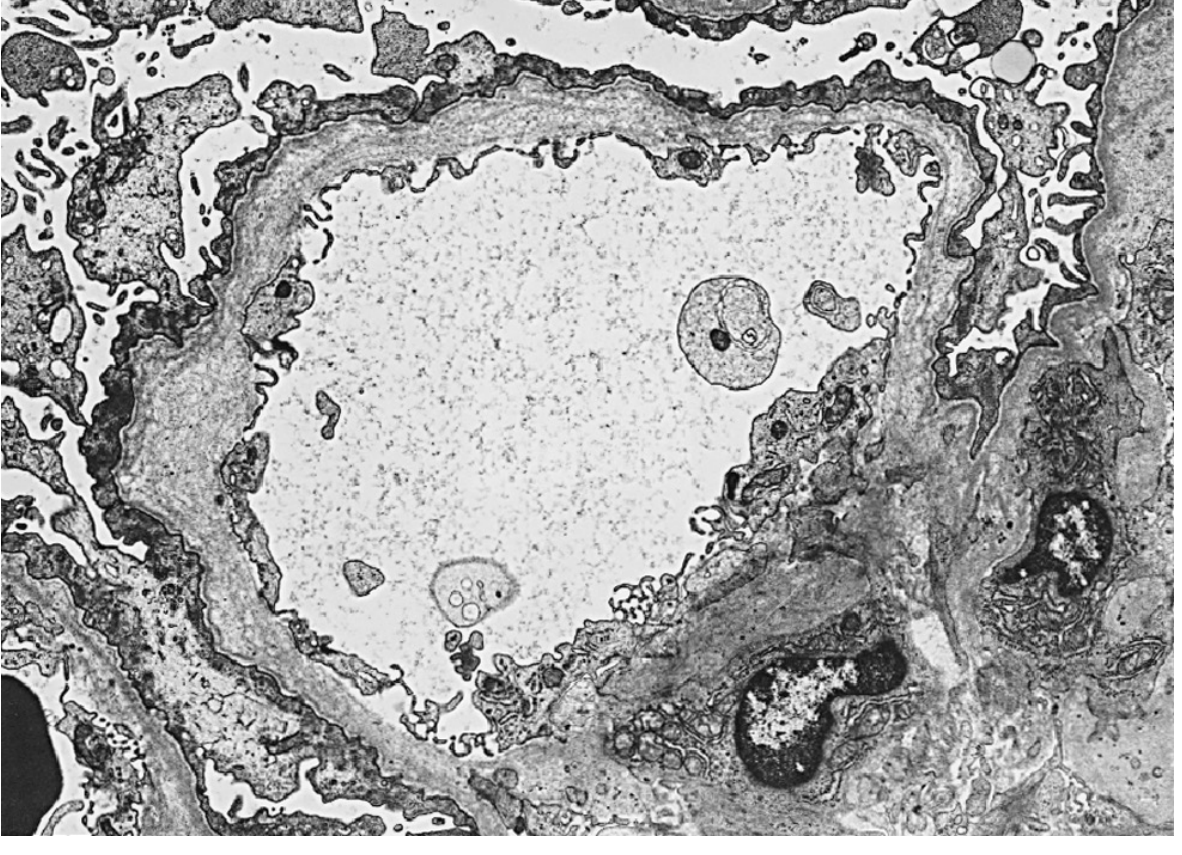
The hallmark findings on EM (progression over time):
- Early: GBM thinning (similar to thin basement membrane nephropathy)
- Later: GBM thickening → irregular multilamellation of the lamina densa surrounding electron-lucent areas with electron-dense granules
- Classic: "basket weave" or "split and splintered" appearance - pathognomonic
Areas of thinning, thickening, and splitting coexist in the same biopsy.
Diagnosis
The 2024 ERKNet/ERA/ESPN guideline (Torra et al., Nephrol Dial Transplant 2025) now places genetic testing as the key first-line diagnostic test. It should be performed in anyone with:
- Persistent hematuria of unknown origin
- Proteinuria or kidney failure of unknown cause
- FSGS of unknown origin
- Family history of hereditary nephritis
Diagnostic approach:
| Test | Finding |
|---|---|
| Genetic testing (NGS panel: COL4A3/A4/A5) | Pathogenic variant confirms diagnosis; also determines inheritance pattern |
| Skin biopsy (IHC for α5(IV)) | Absence of α5 is diagnostic of X-linked AS; avoids kidney biopsy in some males |
| Kidney biopsy (EM + IHC) | Required for AR/AD forms; shows basket weave GBM + absent collagen chains |
| Audiogram | High-frequency sensorineural hearing loss |
| Slit lamp exam | Anterior lenticonus |
| Urinalysis | Dysmorphic RBCs, RBC casts; proteinuria |
| ABG/serum creatinine | Assess kidney function |
Clinical tip: absence of extrarenal symptoms does NOT rule out the diagnosis.
Treatment
Renoprotective therapy (first-line, per 2024 guidelines)
ACE inhibitors (ACEi) are standard of care:
- Significantly reduce proteinuria and delay progression to ESKD
- Early initiation provides the greatest benefit - recommended even in patients with microalbuminuria/hematuria alone in high-risk genotypes
- Benefit extends to heterozygous carriers with significant mutations
SGLT2 inhibitors:
- The 2024 ERKNet/ERA/ESPN guideline recommends adding SGLT2i in adults with proteinuria and CKD
- Emerging evidence supports their nephroprotective role in AS
ARBs: Used as alternatives or in combination; additional benefit in animal models.
Oxygen alone / isolated therapies to avoid: Treating hypertension adequately is also important.
Emerging/investigational
- Anti-miR-21 (microRNA inhibitor) - clinical trials ongoing
- Bardoxolone methyl (Nrf2 activator)
- TGF-β inhibition, endothelin A antagonism (animal models)
Kidney transplantation
- Good long-term outcomes overall
- Risk: de novo anti-GBM nephritis in the allograft (because the donor kidney contains α3α4α5(IV) chains absent in the host → treated as foreign antigen)
- Anti-GBM antibodies detectable in a significant proportion but overt nephritis occurs in <3% of cases
- Important: relatives with heterozygous COL4A3/A4/A5 variants should be the last resort for living kidney donation (per 2024 guidelines), given their own risk of progression
Key Distinguishing Points
| Feature | Alport Syndrome | Thin BM Nephropathy |
|---|---|---|
| Genetics | COL4A3/A4/A5 mutations | COL4A3/A4 heterozygous (mild) |
| GBM on EM | Basket weave / splitting | Uniform thinning only |
| Extrarenal features | Deafness, lenticonus | Absent |
| Progression to ESKD | Common (especially males) | Rare |
| IHC (α3/α4/α5) | Absent from GBM | Normal |
⚠️ 2024 Guideline Update - The ERKNet/ERA/ESPN 2024 guideline (PMID 39673454) now formally recommends: (1) genetic testing as the first diagnostic step; (2) early RAS blockade even before overt proteinuria in high-risk patients; (3) SGLT2i added in adults with CKD+proteinuria; and (4) caution around using heterozygous relatives as living kidney donors.
Sources: Brenner & Rector's The Kidney 2-Volume Set; Comprehensive Clinical Nephrology 7e; Harrison's Principles of Internal Medicine 22e; NKF Primer on Kidney Diseases 8e; Torra et al. ERKNet/ERA/ESPN 2024 Practice Guideline, PMID 39673454
Robertsonian Translocation
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Reading File
Loading Image
I have everything I need. Here is the complete answer:
Robertsonian Translocation
Definition
A Robertsonian translocation (ROB) is a structural chromosomal rearrangement in which two acrocentric chromosomes break at or near their centromeres and fuse at their long arms (q arms), with loss of the short arms. The result is a single large derivative chromosome composed of the long arms of both original chromosomes.
- The total chromosome count drops from 46 to 45
- The karyotype is considered functionally balanced despite having only 45 chromosomes, because the lost short arms contain only ribosomal RNA genes (with hundreds of backup copies on the other acrocentric chromosomes) - their loss causes no clinical harm
- The resulting translocation chromosome is typically pseudodicentric (has two centromeric regions fused near each other)
Chromosomes Involved
Only the five pairs of acrocentric chromosomes - those with very short, satellite-bearing p arms - can participate in Robertsonian translocations:
| Group | Chromosomes |
|---|---|
| Group D | 13, 14, 15 |
| Group G | 21, 22 |
Any combination of one Group D and one Group G chromosome (or within the same group) can fuse. The most common Robertsonian translocations are:
| Translocation | Notation | Frequency |
|---|---|---|
| rob(13;14) | t(13q;14q) | ~1 in 1,300 persons - most common chromosome rearrangement in humans |
| rob(14;21) | t(14q;21q) | Most clinically significant (risk of Down syndrome) |
| rob(13;21), rob(15;21), rob(21;22) | Various | Less common |
| rob(21;21) | t(21q;21q) | Rare, but critical - 100% recurrence risk |
Overall prevalence of Robertsonian translocations in the general population is approximately 1 in 1,000.
Structure - What Happens
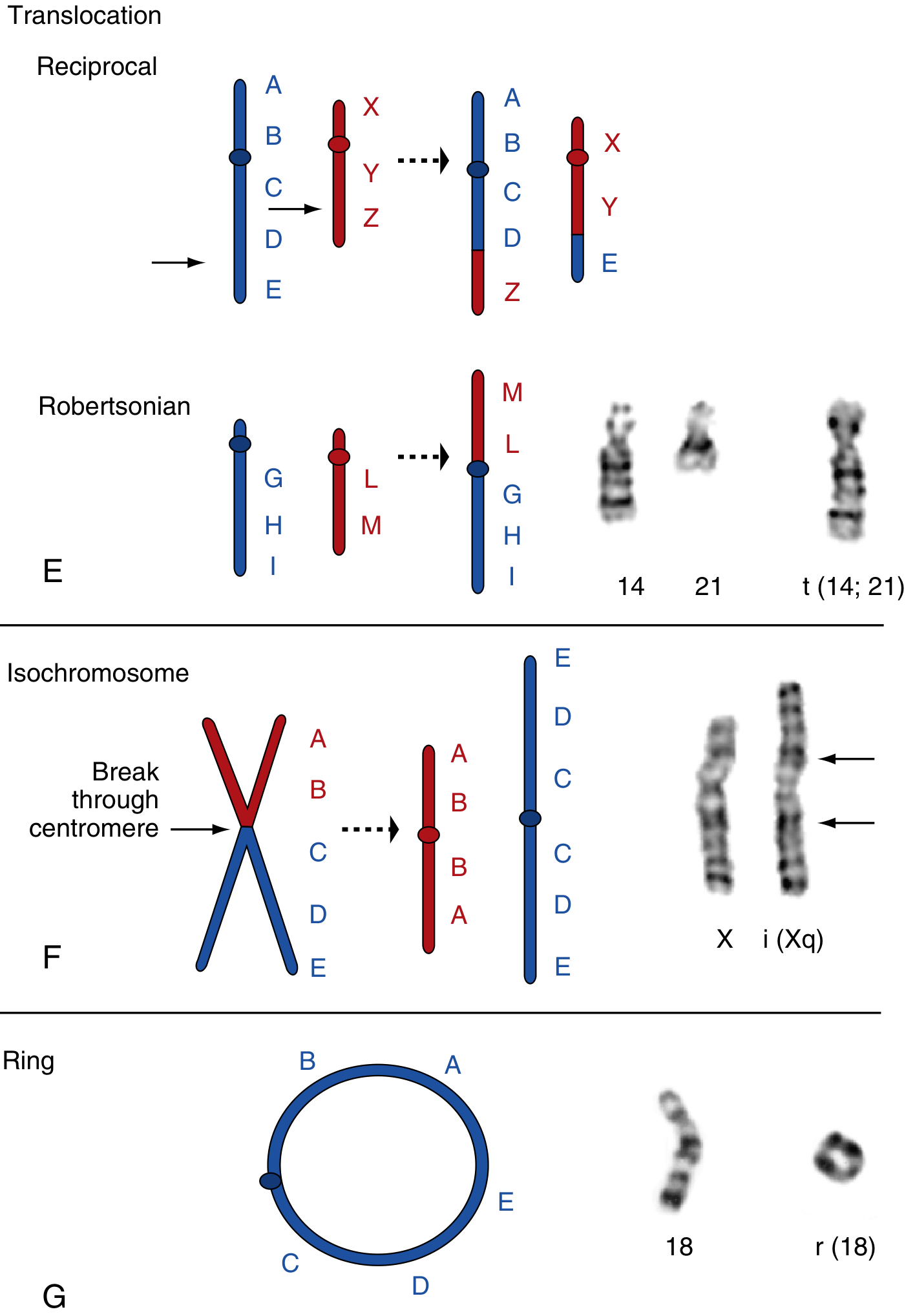
As shown above (panel E), chromosomes 14 and 21 each contribute their long arms to form the single translocation chromosome t(14;21) - physically verified by G-banding on the right. The short arms are lost.
Carrier Phenotype
A balanced Robertsonian translocation carrier:
- Has 45 chromosomes (not 46)
- Is phenotypically normal - no clinical manifestations
- BUT carries significant risk of unbalanced offspring (see below)
Rare individuals with two copies of the same Robertsonian translocation (e.g., two copies of rob(13;14)) have only 44 chromosomes, are phenotypically normal, and lack any normal copies of the acrocentrics involved.
Meiotic Segregation and Gamete Outcomes
This is the critical clinical concept. During meiosis, the three chromosomes involved (two normal acrocentrics + one translocation chromosome) must align together, forming a trivalent (for Robertsonian translocations) rather than the quadrivalent seen in reciprocal translocations.
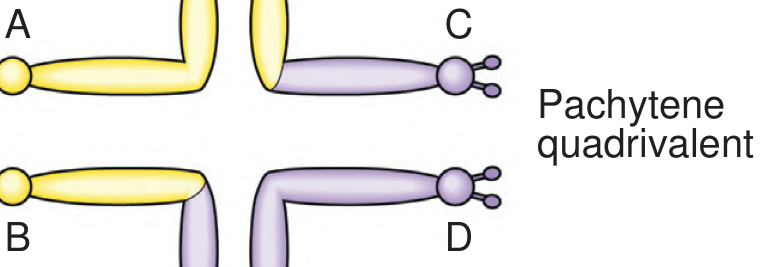
For a rob(14;21) carrier, the six possible gametes are:
| Gamete | Chromosomes Received | After Fertilization | Outcome |
|---|---|---|---|
| 1 | Normal 14 + Normal 21 | Normal karyotype (46) | Normal child |
| 2 | Translocation t(14;21) only | Balanced carrier (45) | Balanced carrier |
| 3 | t(14;21) + Normal 21 | 46 chromosomes, but 3 copies of 21q | Translocation Down syndrome |
| 4 | Normal 14 only | Monosomy 21 | Lethal (miscarriage) |
| 5 | Normal 21 only | Monosomy 14 | Lethal (miscarriage) |
| 6 | t(14;21) + Normal 14 | Trisomy 14 | Lethal (miscarriage) |
In practice, 1/3 of viable gametes are normal, 1/3 are balanced carriers, and 1/3 lead to unbalanced offspring. The lethal combinations are lost as miscarriages, enriching the viable births for the three "normal/balanced/Down" outcomes.
Recurrence Risks
The empirical recurrence risks depend on which translocation and critically, which parent is the carrier:
| Carrier Parent | rob(14;21) | rob(13;21) / rob(15;21) | rob(21;21) |
|---|---|---|---|
| Mother | ~10% risk of Down syndrome offspring | ~10% | 100% (all offspring have Down syndrome or monosomy 21) |
| Father | ~1-3% risk | ~1-3% | 100% |
The lower risk in male carriers is thought to reflect selection against unbalanced spermatozoa. Female carriers consistently have higher empirical risk because selection is less effective.
The 21q;21q Translocation - Special Case
This is one of the most critical situations in clinical genetics. All gametes from a rob(21;21) carrier are either nullisomic or disomic for chromosome 21. Therefore:
- Every surviving pregnancy = Down syndrome (trisomy 21)
- Every non-surviving pregnancy = monosomy 21 (lethal)
- This is one of the very rare situations with >50% recurrence risk for abnormality
Clinical Significance
1. Translocation Down Syndrome
- Accounts for ~4% of all Down syndrome cases
- Patient has 46 chromosomes (not 47), but is trisomic for all genes on 21q
- Clinically identical to standard trisomy 21
- Key differences from standard trisomy 21:
| Feature | Standard Trisomy 21 | Translocation Down Syndrome |
|---|---|---|
| Chromosome count | 47 | 46 |
| Maternal age effect | Yes (strong) | No |
| Mechanism | Meiotic nondisjunction | Inherited translocation |
| Recurrence risk | Low (~1%) | High (up to 10-15% if parent is carrier) |
| Parental karyotyping needed | No | Yes - essential |
- In ~2/3 of cases, the translocation is de novo in the child (low recurrence risk)
- In ~1/3 of cases, one parent is a balanced carrier (high recurrence risk) - this family requires "translocation tracing" to identify all at-risk relatives
2. Recurrent Pregnancy Loss (RPL)
- Balanced translocations are the most common chromosomal cause of RPL
- Found in 3-5% of couples with RPL vs. 0.2-0.7% in the general population
- Robertsonian translocations account for ~40% of these balanced translocations (reciprocal account for ~60%)
- Unbalanced gametes lead to trisomies or monosomies → miscarriage
3. Infertility
- Unbalanced segregation products predominantly result in miscarriage, reducing effective fertility
Karyotype Notation
Standard ISCN notation examples:
- Balanced carrier (male): 45,XY,rob(14;21)(q10;q10)
- Down syndrome with translocation: 46,XY,rob(14;21)(q10;q10),+21
- rob(21;21) carrier: 45,XX,rob(21;21)(q10;q10)
Management / Genetic Counseling Points
- Always karyotype a child with Down syndrome - not just to confirm diagnosis but to detect translocation vs. free trisomy
- If translocation is found in the proband, karyotype both parents and then extend to relevant relatives ("translocation tracing / chasing")
- Carriers should receive counseling about recurrence risk based on specific translocation and sex of carrier parent
- Prenatal diagnosis (CVS or amniocentesis) is indicated in subsequent pregnancies of identified carriers
- For rob(21;21) carriers - all pregnancies are at risk; preimplantation genetic testing (PGT) can be discussed as a route to unaffected offspring
- Balanced carrier relatives with heterozygous Robertsonian translocations - consider implications for living kidney donation if co-existing renal conditions exist (per recent hereditary renal disease guidelines)
Sources: Thompson & Thompson Genetics and Genomics in Medicine, 9th edition; Emery's Elements of Medical Genetics and Genomics; Creasy & Resnik's Maternal-Fetal Medicine; Henry's Clinical Diagnosis and Management by Laboratory Methods; Basic Medical Biochemistry 6e
Turner syndrome
Finding Sources
Searching PubMed
Turner syndrome management guidelines
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive information from multiple authoritative sources. Here is the complete answer:
Turner Syndrome
Definition & Epidemiology
Turner syndrome (TS) results from complete or partial monosomy of the X chromosome and is characterized by primary hypogonadism in phenotypic females.
- Most common sex chromosome abnormality in females
- Incidence: approximately 1 in 2,000-3,000 live-born females
- Important: 45,X is the most common cytogenetic abnormality in spontaneously aborted fetuses - it accounts for ~18% of all chromosomally abnormal miscarriages. Only ~1% of monosomy X embryos survive to birth.
Karyotypes
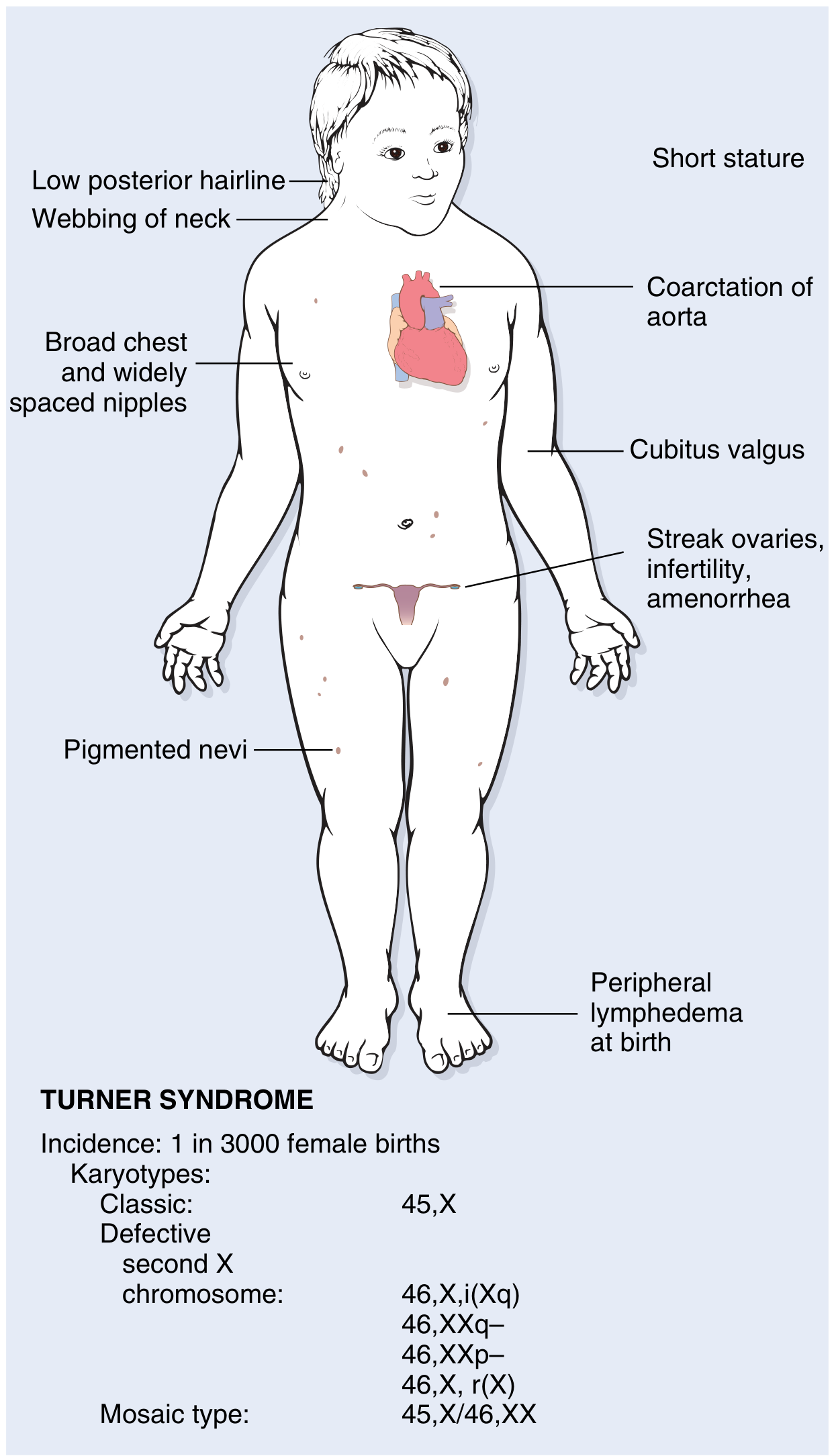
Three categories of karyotypic abnormality are recognized:
1. Classic monosomy X (57%)
- 45,X - complete loss of one X chromosome
- Most severe phenotype
2. Structural abnormalities of the X chromosome (14%)
In order of frequency:
| Karyotype | Description |
|---|---|
| 46,X,i(X)(q10) | Isochromosome of the long arm - loss of the entire short arm; most common structural variant |
| 46,X,r(X) | Ring chromosome - deletion of both short and long arm ends |
| 46,X,del(Xp) | Deletion of short arm |
| 46,X,del(Xq) | Deletion of long arm |
All produce partial monosomy of X.
3. Mosaicism (29%)
- A 45,X cell line coexists with one or more other cell populations
- Common mosaic karyotypes:
- 45,X / 46,XX (mildest - may have near-normal phenotype, primary amenorrhea only)
- 45,X / 46,XY (mixed gonadal dysgenesis - risk of gonadoblastoma)
- 45,X / 47,XXX
- 45,X / 46,X,i(X)(q10)
- Note: With sensitive molecular techniques, mosaicism may be present in up to 75% of cases - conventional cytogenetics underestimates this
Y chromosome material (5-10% of TS patients): Present as complete Y or Y fragments translocated onto other chromosomes → significantly increased risk of gonadoblastoma → traditionally indicated gonadectomy
Origin of the Missing Chromosome
- In ~75-80% of cases, the retained X chromosome is maternal in origin
- This means the error is in paternal gametogenesis (the paternal sex chromosome, either X or Y, is lost)
- No maternal age effect (unlike autosomal trisomies)
Pathogenesis
Streak Ovaries - Key Mechanism
Both X chromosomes are required for normal ovarian development. In Turner syndrome:
- Fetal ovaries develop normally during the first 18 weeks of gestation
- After week 18, absence of the second X leads to accelerated, irreversible oocyte loss, complete by age 2 years
- Result: ovaries become white fibrous streak gonads - devoid of ova and follicles
- Described memorably as: "menopause occurs before menarche"
SHOX Gene Haploinsufficiency
The SHOX gene (short stature homeobox) at Xp22.33 lies in the pseudoautosomal region (PAR1) and normally escapes X-inactivation - so both males and females have two copies. In Turner syndrome:
- SHOX haploinsufficiency → short stature (the primary molecular explanation for growth failure)
- SHOX is expressed in growth plates of long bones (radius, ulna, tibia, fibula)
- SHOX deletions also explain short stature in 2-5% of otherwise normal short children
- Conversely, excess SHOX copies in Klinefelter syndrome (47,XXY) → tall stature
Genes on the short arm (Xp) primarily determine somatic features (cardiac, renal, skeletal), while genes on the long arm (Xq) mainly affect fertility and menstruation.
Clinical Features
Neonatal / Infancy
- Peripheral lymphedema of the dorsum of hands and feet (lymph stasis) - a presenting sign
- Neck swelling due to dilated lymphatics → cystic hygroma → resolves leaving webbing
- Congenital heart disease detected early
Classic Features (Adolescents / Adults)
| System | Features |
|---|---|
| Growth | Short stature - final height rarely exceeds 150 cm untreated; begins in early childhood |
| Neck | Webbing of the neck (pterygium colli); low posterior hairline |
| Chest | Broad "shield chest" with widely spaced nipples |
| Limbs | Cubitus valgus (increased carrying angle of arms); short 4th metacarpal; lymphedema |
| Reproductive | Primary amenorrhea (TS is the single most common cause, ~1/3 of all cases); streak ovaries; infantile genitalia; minimal breast development; infertility |
| Cardiovascular | Bicuspid aortic valve (30-50%); preductal coarctation of aorta (30%); aortic root dilation (30%); 100-fold increased risk of aortic dissection; ~5% of girls with coarctation have Turner syndrome |
| Renal | Horseshoe kidney; duplicated collecting system; malrotation (~30%) |
| Skin | Multiple pigmented nevi |
| Facial | High-arched palate; micrognathia; low-set ears; epicanthal folds |
| Neurological/cognitive | Intelligence usually normal; subtle defects in non-verbal, visual-spatial processing |
| Endocrine | Autoimmune thyroiditis → hypothyroidism in ~50% (especially with isochromosome Xq); glucose intolerance; insulin resistance; metabolic syndrome in a subset |
| GI | Celiac disease; nonalcoholic fatty liver disease |
| Hearing | Sensorineural and/or conductive hearing loss |
Diagnosis
| Tool | Finding |
|---|---|
| Karyotype (gold standard) | 45,X or mosaic/structural variant |
| Prenatal ultrasound | Cystic hygroma, hydrops, cardiac abnormalities, shortened femur |
| NIPT (cell-free DNA) | Can detect monosomy X prenatally |
| Hormones (post-puberty) | Elevated FSH and LH (hypergonadotropic hypogonadism); low/absent estrogen |
| Pelvic ultrasound | Streak ovaries; small/absent uterus |
| Echocardiography | Bicuspid aortic valve, coarctation, aortic root dimensions |
| Renal ultrasound | Structural anomalies in ~30% |
Key clinical trigger: Short stature + primary amenorrhea in an adolescent girl = Turner syndrome until proven otherwise.
Management (Multidisciplinary)
Growth - Recombinant Growth Hormone (rGH)
- Started early (before age 5-6 ideally) to maximize adult height gain
- Without treatment, final height rarely exceeds 150 cm
- Caveat: rGH worsens insulin resistance - monitor glucose
Hormone Replacement Therapy (HRT)
- Estrogen initiated at age-appropriate time (~11 years) to induce puberty:
- Breast and uterine development
- Supports linear growth
- Maintains bone mineralization (prevents osteoporosis)
- Doses increased gradually over 2-4 years
- Progestins added later for withdrawal bleeds (endometrial protection)
- HRT maintained through normal menopausal age (~50 years)
Fertility
- Majority infertile; spontaneous pregnancy rare
- Successful pregnancy achieved via ovum donation + IVF in some women
- Risk: cardiac complications (aortic dissection risk is high during pregnancy) - requires expert counseling and careful cardiac monitoring
Cardiovascular Surveillance
- Regular cardiology follow-up throughout life
- Serial MRI of aortic root dimensions (preferred over echo for aorta)
- Antibiotic prophylaxis for dental/surgical procedures
- Aggressive blood pressure management
- Pregnancy contraindicated or very high-risk in those with aortic root dilation
Gonadoblastoma Risk (Y material)
- If Y chromosome sequences identified → gonadectomy traditionally recommended
- Modern approach: individualize decision weighing gonadal function potential and patient autonomy
- Intra-abdominal dysgenetic gonads carry up to 35% risk of germ cell cancer
Surveillance Schedule
| Domain | Monitoring |
|---|---|
| Cardiac | Echo/MRI aorta; BP at every visit |
| Renal | Ultrasound; BP; UTI screening |
| Thyroid | Annual TSH |
| Metabolic | Weight, glucose tolerance, lipids |
| Bone | DEXA scan |
| Hearing | Audiogram every 3-5 years |
| Eyes | Ophthalmology |
| Liver | LFTs, liver ultrasound |
| Celiac | Anti-tTG antibodies |
| Cognitive/educational | Neuropsychological assessment |
Key Comparisons
| Feature | Turner (45,X) | Noonan Syndrome |
|---|---|---|
| Sex | Females only | Males and females |
| Karyotype | Abnormal (monosomy X) | Normal (PTPN11 mutation) |
| Short stature | Yes | Yes |
| Webbed neck | Yes | Yes |
| Cardiac | Left-sided (coarctation, bicuspid Ao valve) | Right-sided (pulmonary stenosis, HCM) |
| Ovarian function | Streak ovaries | Often normal |
| Inheritance | Sporadic | Autosomal dominant |
Noonan syndrome is often called the "male Turner" but is a distinct RASopathy - the physical similarity is coincidental.
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Robbins & Kumar Basic Pathology; Harrison's Principles of Internal Medicine 22e (2025); The Developing Human: Clinically Oriented Embryology; Creasy & Resnik's Maternal-Fetal Medicine
This is a shared conversation. Sign in to Orris to start your own chat.