Short stature in detail
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Searching PubMed
"short stature" children diagnosis management
Loading Image
Searching Images
short stature causes classification children growth chart
Now I have comprehensive information from multiple authoritative sources. Let me compile the detailed response.
Short Stature — A Comprehensive Overview
Definition
Short stature is defined as a height more than 2 standard deviations (SD) below the mean for age and sex, corresponding to below the 3rd percentile on growth charts — affecting approximately 2.5% of children.
Growth failure is distinct and defined as:
- Height <2 SD below mid-parental height, OR
- Height velocity <10th percentile for age resulting in downward crossing of height percentiles
The majority of children with short stature are healthy; true growth failure is typically pathologic and requires evaluation. — The Harriet Lane Handbook, 23rd ed.
Mid-Parental Height Calculation
Predicted adult height is calculated as follows (Rogol, 2004):
- Boys: [Father's height + (Mother's height + 13 cm)] ÷ 2
- Girls: [(Father's height − 13 cm) + Mother's height] ÷ 2
Final height can also be predicted by adding 6.5 cm (boys) or subtracting 6.5 cm (girls) from the midparental height. — Harrison's Principles of Internal Medicine, 22nd ed.
Classification of Causes
1. Non-Pathologic (Most Common)
A. Familial Short Stature (Most Common Overall)
- Parents or close relatives are short
- Normal birth weight and length, growth rate declines in first 2–3 years then parallels but falls below 5th percentile
- Bone age = chronologic age (normal)
- Growth velocity: normal
- Puberty at normal age
- Adult height is short, near mid-parental height
- FDA-approved recombinant GH for idiopathic short stature can increase predicted height by >7 cm
B. Constitutional Growth Delay (2nd Most Common)
- Majority are boys with normal birth length/weight
- Growth decelerates in first 2 years, then returns to normal velocity but at a lower percentile
- Bone age = height age (both delayed relative to chronologic age)
- Delayed puberty — later than peers
- Family history of delayed growth and puberty common
- Adult height is ultimately normal
- Differentiated from familial short stature by bone age: delayed in constitutional delay, normal in familial short stature
Comparison Table (Pathologic vs Non-Pathologic):
| Feature | Familial Short Stature | Constitutional Growth Delay | Pathologic |
|---|---|---|---|
| Growth velocity | Normal | Normal | Decreased |
| Onset of puberty | Normal | Delayed | Depends on cause |
| Family history | Short parents | Delayed puberty | +/− |
| Bone age | Normal | Delayed | Usually delayed |
| Adult height | Short (near mid-parental) | Normal | Depends on cause |
2. Pathologic Causes
A. Endocrine Disorders
Growth Hormone Deficiency (GHD)
- ~15% of children evaluated for growth retardation have an endocrine disorder; ~half of those (~8% of all short stature) have GHD
- Characteristics: short stature, low growth velocity, immature facial appearance, delayed bone age, increased adiposity
- Congenital GHD: size at birth is usually normal (in utero IGF-1 is not GH-dependent); micropenis may be present in boys
- Adults with GHD: reduced muscle mass (sarcopenia), increased central adiposity, osteoporosis, dyslipidemia, increased cardiovascular risk
- Causes: hypothalamic disease, disruption of portal blood supply, GHRHR mutations, somatotroph disease
- Can be isolated GHD or part of multiple pituitary hormone deficiency (MPHD) — Tietz Textbook of Laboratory Medicine, 7th ed.
Hypothyroidism
- Growth retardation in children; bone age delayed
Cushing's Syndrome
- Bone age less than chronologic age; decreased growth rate
Precocious Puberty (eventual short adult)
- Bone age greater than chronologic age; initially accelerated growth but premature epiphyseal fusion leads to short final height
B. Chromosomal / Genetic Causes
Turner Syndrome (45,XO)
- Short stature is a universal feature of non-mosaic Turner syndrome
- Loss of SHOX gene (short stature homeobox gene on Xp22.3) accounts for the growth deficit
- Other features: webbed neck, primary amenorrhea, broad chest, widely spaced nipples
- Treatment: GH (often combined with oxandrolone), then estrogen replacement
SHOX Gene Mutations/Deficiency
- SHOX mutations alone cause Leri-Weill dyschondrosteosis (forearm bowing, Madelung deformity)
- 5–15% of children with idiopathic short stature have a SHOX mutation (Xp23.33)
- Forearm anomalies become more evident during puberty
- FDA-approved indication for GH therapy
Down Syndrome (Trisomy 21), Noonan Syndrome, Prader-Willi Syndrome, Russell-Silver Syndrome, Spondyloepiphyseal Dysplasia, Skeletal Dysplasias (achondroplasia, hypochondroplasia)
C. Chronic Systemic Disease
- Celiac disease, Crohn's disease, chronic renal failure, cystic fibrosis, cardiac failure, HIV, liver disease, chronic anemias
- Mechanism: secondary GH receptor resistance; proinflammatory cytokines block GH-mediated signal transduction
- These children show normal or elevated GH with low IGF-1
D. Nutritional / Psychosocial
- Nutritional short stature: caloric deprivation, uncontrolled diabetes, chronic renal failure — GH normal/elevated, IGF-1 low
- Psychosocial short stature: emotional/social deprivation → growth retardation, delayed speech, discordant hyperphagia, attenuated GH response. A nurturing environment restores growth. — Harrison's, 22nd ed.
E. Intrauterine Growth Restriction (IUGR) / Small for Gestational Age (SGA)
- Prenatal-onset growth failure
Bone Age Relationship to Causes
| Bone Age vs Chronologic Age | Growth Rate | Condition |
|---|---|---|
| Bone age < chronologic age | Normal/slightly decreased | Constitutional growth delay |
| Bone age < chronologic age | Decreased | GHD, Cushing's, chronic disease, severe malnutrition, psychosocial deprivation |
| Bone age = chronologic age | Normal/slightly decreased | Familial short stature, skeletal dysplasias |
| Bone age = chronologic age | Decreased | Down syndrome, Turner syndrome |
| Bone age > chronologic age | Initially increased → short adult | Exogenous androgens, precocious puberty |
— Textbook of Family Medicine, 9th ed.
Diagnostic Approach
When to Evaluate
- Height >3 SD below mean, OR
- Growth rate has decelerated (height velocity declining)
- Clinical judgment integrated with auxologic data and family history
- Bone age by left wrist/hand radiograph (reliable after age 2 years)
Diagnostic Algorithm
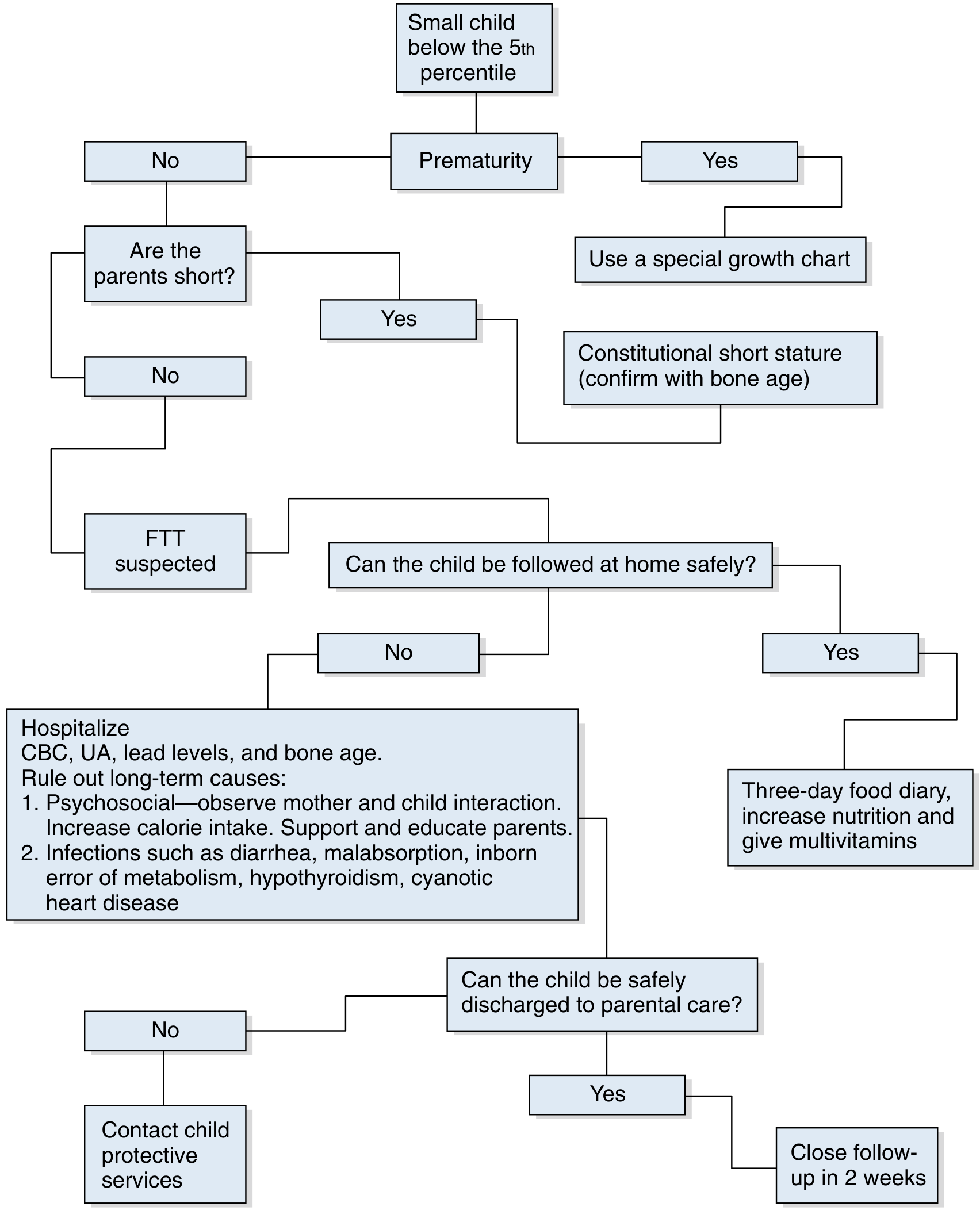
Laboratory Workup
First-line investigations:
- CBC with differential (anemia, malignancy, inflammation)
- CRP/ESR (inflammation, infection)
- CMP — renal and liver function, calcium (malnutrition, renal/liver disorders)
- Urinalysis
- TSH, free T4 (hypothyroidism)
- TTG + IgA (celiac disease)
- Bone age radiograph
Second-line / targeted:
- IGF-1 and IGFBP-3 — proxy for GH axis; IGFBP-3 has higher specificity in children <10 years
- Levels below the 5th percentile suggest GH deficiency
- Karyotype or SHOX gene testing — for Turner syndrome and SHOX mutations (especially girls with unexplained short stature)
- GH stimulation testing (provocative):
- GH deficiency is best assessed by provocative tests (exercise, insulin-induced hypoglycemia, arginine, L-dopa, GHRH, macimorelin)
- Normal response: GH peak >7 μg/L in children; a single subnormal test should be confirmed with a second test
- Insulin Tolerance Test (ITT): gold standard; requires glucose <40 mg/dL; contraindicated with cardiac disease, seizure disorder
- Arginine stimulation: 0.5 g/kg IV over 30 min; peak GH should be ≥5–7 ng/mL
- Random GH measurements do NOT distinguish GHD from normal
- Pituitary MRI — for mass lesions or structural defects when GHD suspected
- Specific tests for cystic fibrosis, HIV, tuberculosis as clinically indicated
Growth Velocity Thresholds
| Normal growth velocity | Short stature with NORMAL velocity |
|---|---|
| ≥4–5 cm/year | → Familial, primordial, or constitutional delay |
| <4–5 cm/year | → Genetic short stature, chronic illness, malnutrition, Turner, endocrine disorders |
— Tietz Textbook of Laboratory Medicine, 7th ed.
Treatment
FDA-Approved Indications for Recombinant GH (Somatotropin)
- Growth hormone deficiency — children and adults
- Turner syndrome — GH (often + oxandrolone) to increase height
- Chronic renal failure
- Prader-Willi syndrome
- Small for gestational age (SGA) — if not caught up by age 2–4 years
- SHOX deficiency
- Idiopathic short stature — height <2.25 SD below mean and unlikely to attain normal adult height
Pediatric dosing: 0.02–0.05 mg/kg/day SC
Adult GH replacement: 0.1–1.25 mg SC daily (for confirmed GHD)
Laron dwarfism (GH receptor deficiency/GH resistance): GH levels are elevated; treatment with GH is ineffective — requires IGF-1 replacement (mecasermin)
For Specific Conditions
- Hypothyroidism → L-thyroxine
- Cushing's syndrome → treatment of underlying cause
- Psychosocial short stature → nurturing environment restores growth
- Nutritional short stature → caloric rehabilitation
- Turner syndrome → GH + oxandrolone; estrogen replacement for pubertal induction
- Precocious puberty → GnRH analogs to slow bone maturation
Key Differentiating Points (Exam Summary)
| Feature | Familial SS | Constitutional Delay | GHD | Turner |
|---|---|---|---|---|
| Bone age | Normal | Delayed | Delayed | Normal or delayed |
| Growth velocity | Normal | Normal | Low | Low |
| Puberty | Normal | Delayed | Depends | Absent (primary amenorrhea) |
| IGF-1 | Normal | Normal | Low | Normal |
| Karyotype | Normal | Normal | Normal | 45,XO |
| Adult height | Short | Normal | Short (if untreated) | Short |
Sources:
- The Harriet Lane Handbook, 23rd ed. (Johns Hopkins)
- Harrison's Principles of Internal Medicine, 22nd ed.
- Tietz Textbook of Laboratory Medicine, 7th ed.
- Textbook of Family Medicine, 9th ed.
- Swanson's Family Medicine Review
Recent evidence: A 2026 systematic review (PMID 41723458 — Mameli et al., Ital J Pediatr) specifically addresses the clinical and biochemical evaluation of children with short stature in primary care settings — consistent with the approach outlined above.
This is a shared conversation. Sign in to Orris to start your own chat.