Ischemic heart disease
Reading File
Finding Sources
Finding Sources
Searching PubMed
"ischemic heart disease"[MeSH Terms] AND "management"[MeSH Terms]
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
I now have all the essential material for a comprehensive answer. Here is the full structured overview of Ischemic Heart Disease.
Ischemic Heart Disease (IHD)
Definition
IHD is a broad term encompassing several overlapping syndromes caused by an imbalance between myocardial oxygen demand and coronary blood supply (perfusion). In more than 90% of cases, it results from coronary artery atherosclerosis, and IHD is used virtually synonymously with coronary artery disease (CAD). It remains the leading cause of mortality worldwide, accounting for approximately 7.5 million deaths per year.
- Robbins & Kumar Basic Pathology, p. 351
Clinical Syndromes of IHD
IHD presents across a spectrum of four overlapping syndromes:
| Syndrome | Mechanism | Key Feature |
|---|---|---|
| Angina Pectoris | Ischemia without myocyte death | Reversible chest pain |
| Myocardial Infarction (MI) | Ischemia severe/prolonged enough to cause myocyte necrosis | Cardiomyocyte death |
| Chronic IHD with CHF | Accumulated ischemic insults - pump failure | Progressive cardiac decompensation |
| Sudden Cardiac Death (SCD) | Ischemia-induced lethal ventricular fibrillation | Death within 1 hour of symptom onset |
The term Acute Coronary Syndrome (ACS) applies to three of these: unstable angina, MI, and SCD.
- Robbins & Kumar Basic Pathology, p. 351-352
Pathogenesis
The Two Core Mechanisms
1. Fixed (Chronic) Atherosclerotic Occlusion
- < 70% luminal obstruction: Asymptomatic even with exertion
- > 70% occlusion ("critical stenosis"): Symptoms on exertion - produces stable angina
- ≥ 90% occlusion: Inadequate flow even at rest - contributes to unstable angina
Clinically significant plaques tend to occur within the first several centimeters of the LAD and LCX takeoff from the aorta, and along the entire length of the RCA. Slowly progressive occlusion can allow collateral vessel formation, protecting against MI even with complete occlusion; acute blockage leaves no time for collaterals, so infarction results.
2. Acute Plaque Change with Superimposed Thrombosis/Vasospasm
This is the mechanism underlying ACS. The initiating event is sudden plaque disruption (rupture or erosion), exposing thrombogenic subendothelial contents, leading to rapid thrombus formation.
Vulnerable plaques are those with:
- Large atheromatous (lipid) cores
- Thin overlying fibrous caps
- High macrophage infiltration (secrete metalloproteinases degrading collagen)
- Few smooth muscle cells (less collagen synthesis)
A critical and sobering insight: two-thirds of ruptured plaques had ≤ 50% stenosis before rupture, and 85% had ≤ 70% stenosis. This means most MIs occur in plaques that were NOT previously causing symptoms.
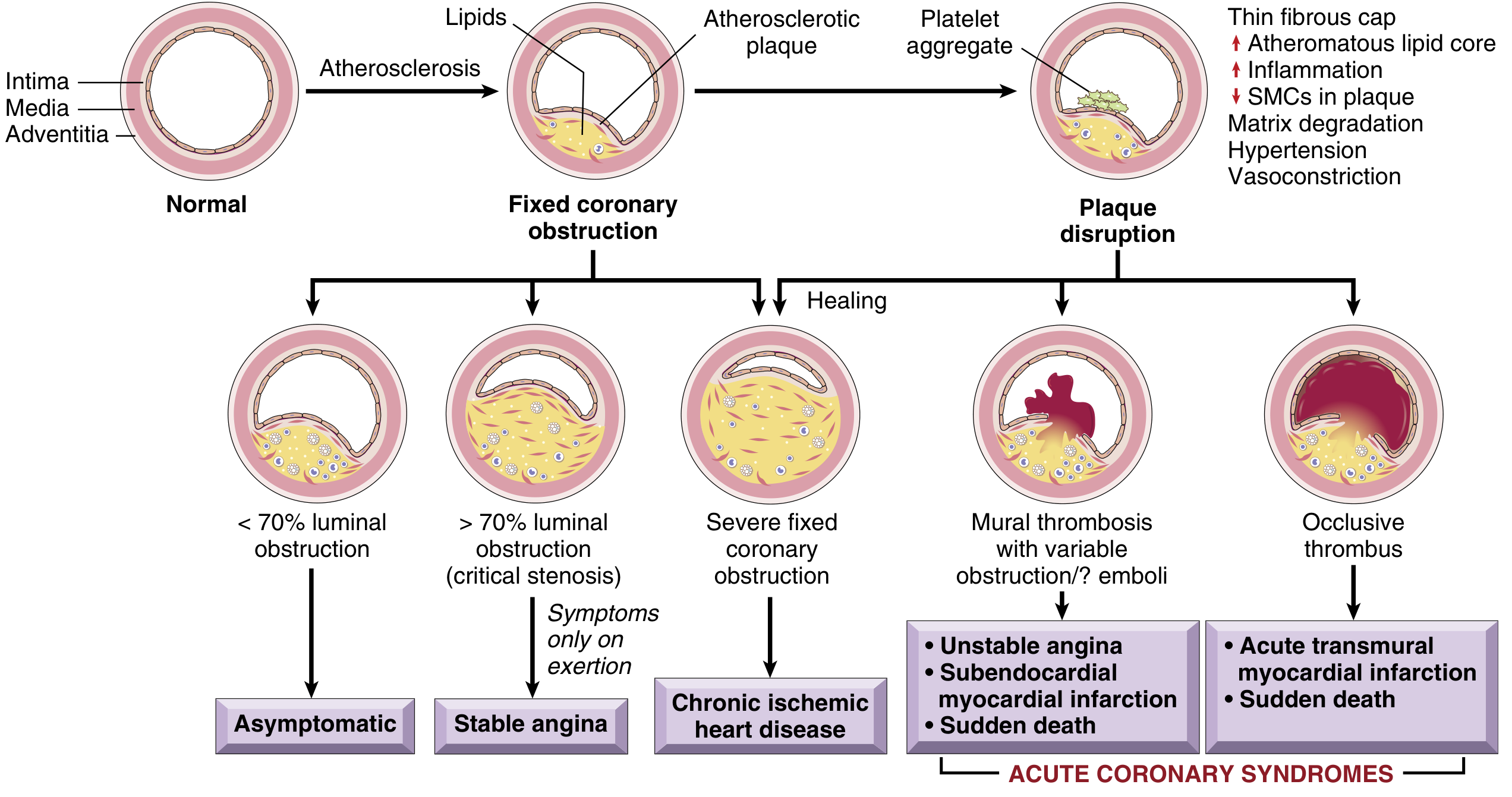
- Robbins & Kumar Basic Pathology, pp. 352-353
Angina Pectoris - Three Types
Angina is intermittent chest discomfort from transient, reversible ischemia - insufficient to cause myocyte necrosis. Mediators released include adenosine, bradykinin, and other molecules stimulating autonomic nerves.
| Type | Mechanism | Relief |
|---|---|---|
| Stable (Typical) Angina | Fixed > 70% stenosis; triggered by exertion or increased demand | Rest or nitroglycerin |
| Prinzmetal (Variant) Angina | Coronary artery spasm, even in non-atherosclerotic vessels | Calcium channel blockers; can occur at rest |
| Unstable Angina | Plaque erosion/rupture + nonocclusive thrombus | Not relieved by rest or nitroglycerin; ACS requiring urgent treatment |
- Robbins & Kumar Basic Pathology, pp. 353-354
Myocardial Infarction
Sequence of Events in a Typical MI
- Plaque erosion or disruption exposes subendothelial collagen and necrotic plaque contents
- Platelets adhere, aggregate, and release thromboxane A2, ADP, and serotonin - causing further aggregation and vasospasm
- Coagulation activation via tissue factor adds to the growing thrombus
- Within minutes, the thrombus may completely occlude the lumen
Angiography within 4 hours of MI shows thrombosis in ~90% of cases. By 12-24 hours (without intervention), this falls to ~60%, as some thrombi lyse spontaneously.
Myocardial Response Timeline
| Time After Occlusion | Effect |
|---|---|
| 1-2 minutes | Loss of myocyte contractile function |
| 20-40 minutes | Irreversible myocyte death begins |
| Hours to days | Gross and histologic changes of infarction develop |
Morphologic Changes in MI - Timeline
| Time | Gross Changes | Histologic Changes |
|---|---|---|
| 0-12 hours | None visible | Early coagulative necrosis; wavy fibers |
| 12-24 hours | Dark mottling | Coagulative necrosis; neutrophil infiltration begins |
| 1-3 days | Mottling with yellow-tan center | Dense neutrophil infiltration |
| 3-7 days | Hyperemic border; yellow-tan softening | Macrophages begin phagocytosing debris |
| 1-2 weeks | Depressed, yellow-tan, soft | Granulation tissue with neovascularization |
| Weeks-months | Gray-white, contracted scar | Dense collagenous scar (fibrosis) |
- Robbins & Kumar Basic Pathology, pp. 354-357
Infarct Location and Affected Vessels
| Occluded Vessel | Territory at Risk |
|---|---|
| LAD (most common) | Anterior LV wall, anterior 2/3 of interventricular septum, apex |
| RCA | Posterior LV wall, posterior 1/3 of septum, RV free wall |
| LCX | Lateral LV wall |
Types of MI
- Transmural (STEMI): Full-thickness necrosis, typically due to complete occlusive thrombus
- Subendocardial (NSTEMI): Inner 1/3-1/2 of myocardium, typically due to partially occlusive thrombus or severe fixed stenosis with increased demand
Complications of MI
| Complication | Timing | Notes |
|---|---|---|
| Arrhythmias | Immediate-hours | Most common cause of early death; V-fib |
| LV pump failure / cardiogenic shock | Hours-days | 10-15% of hospitalized MI patients |
| Free wall rupture | 3-7 days | Peak timing; hemopericardium and tamponade |
| Interventricular septal rupture | 3-7 days | Acute VSD; harsh systolic murmur |
| Papillary muscle rupture | 2-7 days | Acute mitral regurgitation |
| Ventricular aneurysm | Weeks-months | Paradoxical wall motion; risk of mural thrombus |
| Pericarditis (Dressler syndrome) | Days to weeks | Fibrinous or autoimmune |
| Mural thrombus | Days-weeks | Risk of systemic embolism |
- Robbins & Kumar Basic Pathology, pp. 357-360
Risk Factors
Non-modifiable:
- Age (risk rises progressively)
- Male sex (though gap narrows post-menopause in women)
- Family history / genetic predisposition
Modifiable (major):
- Hyperlipidemia (elevated LDL)
- Hypertension
- Smoking
- Diabetes mellitus
- Obesity
- Physical inactivity
Mortality from IHD in the US has declined by ~50% since its peak in 1963, largely due to smoking cessation programs, treatment of hypertension and diabetes, and cholesterol-lowering agents (statins), with a smaller contribution from therapeutic advances.
- Robbins & Kumar Basic Pathology, p. 352; Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 2778
Diagnosis
Troponin (I and T) are the gold-standard biomarkers - cardiac-specific, begin rising within 3-4 hours of MI, peak at 12-24 hours, remain elevated for up to 10-14 days (troponin T). MI is diagnosed by symptoms + ECG changes + abnormal cardiac troponins.
- STEMI: ST elevation in ≥ 2 contiguous leads; requires emergent reperfusion
- NSTEMI: No ST elevation; troponin elevated; may show ST depression or T-wave changes
Management
Angina (Stable)
All patients with IHD should receive guideline-directed medical therapy including lifestyle modification and modifiable risk factor management.
Anti-ischemic drugs:
-
Beta-blockers (first-line): Reduce heart rate and contractility, lowering myocardial O2 demand. Cardioselective beta-1 blockers preferred. Avoid in vasospastic angina.
-
Calcium channel blockers: Arterial vasodilators. First-line for vasospastic (Prinzmetal) angina. Avoid non-dihydropyridines in HFrEF.
-
Organic nitrates (nitroglycerin): Reduce preload and dilate coronary arteries. All angina patients should have sublingual nitroglycerin. Nitrate-free interval required to prevent tolerance.
-
Ranolazine: Sodium channel blocker; reduces late inward Na current, decreasing intracellular Ca overload. Used when other therapies fail; can prolong QT interval.
-
Lippincott Illustrated Reviews: Pharmacology, p. 424
ACS (Unstable Angina / NSTEMI / STEMI)
- Antiplatelet therapy: Aspirin + P2Y12 inhibitor (clopidogrel, ticagrelor, or prasugrel) - dual antiplatelet therapy (DAPT)
- Anticoagulation: Heparin (unfractionated or LMWH), bivalirudin
- Beta-blockers (early)
- Statins (high-intensity)
- ACE inhibitors / ARBs (especially with reduced EF or anterior MI)
Reperfusion Therapy (STEMI)
- Primary PCI (percutaneous coronary intervention): Preferred when available within 90 minutes (door-to-balloon time); most effective at limiting infarct size
- Thrombolysis (fibrinolytics): When PCI is not available within 120 minutes; most effective when given early (< 3 hours)
- CABG (coronary artery bypass grafting): For multivessel or left main disease not amenable to PCI; also used in cardiogenic shock
Early thrombolysis and/or angioplasty can be highly successful in limiting myocardial necrosis, since myocyte death begins at 20-40 minutes and is not complete for several hours.
- Robbins & Kumar Basic Pathology, p. 355; Tintinalli's Emergency Medicine, p. 221
Key Concepts Summary
- IHD = supply-demand mismatch, most often from atherosclerotic CAD
- Most MIs arise from non-critically stenotic plaques that rupture acutely
- The four syndromes: stable angina, unstable angina/NSTEMI, STEMI, SCD
- "Time is muscle" - reperfusion within the first 1-2 hours salvages the most myocardium
- Statins are beneficial beyond lipid-lowering: they stabilize plaques by reducing inflammation and reversing endothelial dysfunction
This is a shared conversation. Sign in to Orris to start your own chat.