Explain me the chapter platelets homeostasis and every topic inside it
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Can yo do it from guton and gnomon
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have all the content needed from both textbooks. Let me compile the full, detailed explanation.
Platelets, Hemostasis, and Blood Coagulation
From Guyton & Hall Textbook of Medical Physiology and Ganong's Review of Medical Physiology
Overview of Hemostasis
Guyton & Hall defines hemostasis as the process that prevents blood loss whenever a vessel is severed or ruptured. It involves four sequential events:
- Vascular constriction
- Formation of the platelet plug (primary hemostasis)
- Blood coagulation (secondary hemostasis)
- Fibrous tissue ingrowth into the clot for permanent vessel repair
Ganong adds that hemostasis is the balance between coagulation and anticoagulation, maintaining blood fluid inside vessels while rapidly sealing breaks when injury occurs.
1. Vascular Constriction (Vascular Spasm)
(Guyton & Hall)
Immediately after a vessel is cut or ruptured, the smooth muscle in the vessel wall contracts. This is caused by:
- Local myogenic spasm - direct muscle response to trauma
- Local autacoid factors released from traumatized tissue, endothelium, and platelets
- Nervous reflexes initiated by pain impulses from injured tissue
The most important vasoconstrictor released by platelets is thromboxane A2 (TxA2). The more severe the injury, the stronger the spasm. Spasm can last minutes to hours, buying time for platelet plug formation and coagulation to occur.
Ganong notes the vasoconstriction is also due to serotonin (5-HT) liberated from adherent platelets - for injured arterioles, the lumen can be temporarily obliterated entirely.
2. Physical and Chemical Characteristics of Platelets
(Guyton & Hall)
Platelets (thrombocytes) are minute discs 1-4 micrometers in diameter. They are formed in the bone marrow from megakaryocytes, which fragment into platelets as they pass through capillaries. Normal platelet count: 150,000 - 450,000/µL.
Although anucleate and unable to reproduce, platelets have rich cytoplasmic contents:
| Component | Function |
|---|---|
| Actin, myosin, and thrombostenin | Contractile proteins enabling clot retraction |
| Endoplasmic reticulum & Golgi apparatus | Synthesize enzymes; store Ca²⁺ |
| Mitochondria | Generate ATP and ADP |
| Enzyme systems | Synthesize prostaglandins (including TxA2) |
| Fibrin-stabilizing factor | Cross-links fibrin during coagulation |
| Platelet-derived growth factor (PDGF) | Stimulates vascular repair and cell growth |
The platelet cell membrane has a glycoprotein coat that:
- Repels normal endothelium (prevents inappropriate clotting)
- Adheres to injured endothelium and exposed subendothelial collagen
- Contains phospholipids (platelet factor 3) that activate multiple coagulation steps
3. Formation of the Platelet Plug (Primary Hemostasis)
(Guyton & Hall)
When a small vessel is cut, the platelet plug alone may seal the hole. It follows three steps:
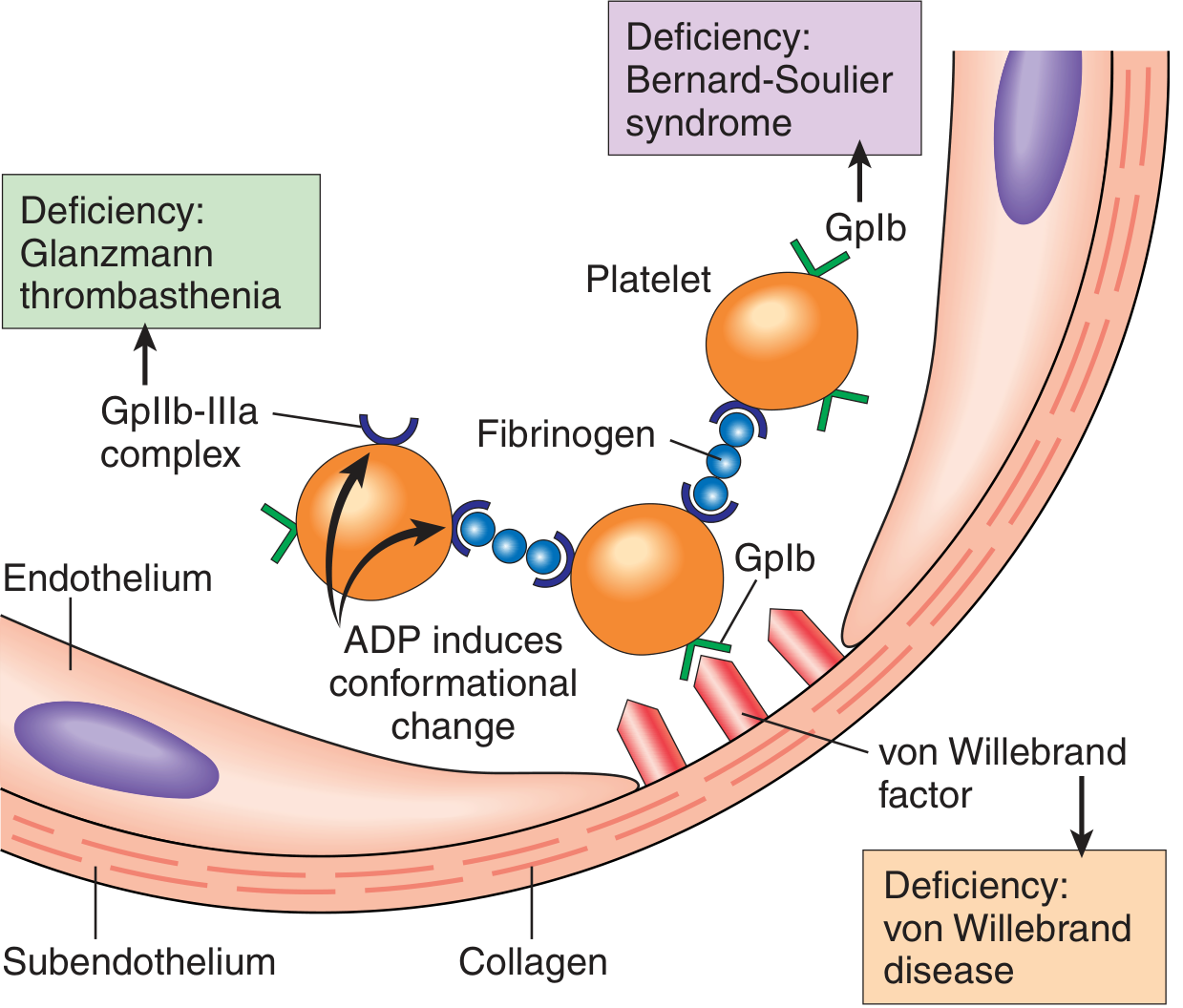
Step 1 - Platelet Adhesion
Platelets contact exposed subendothelial collagen and change their character instantly. von Willebrand factor (vWF) acts as a bridge between collagen and the platelet surface receptor glycoprotein Ib (GpIb). This anchors platelets to the damaged site.
Step 2 - Platelet Activation (Release Reaction)
Adhered platelets swell, form pseudopodia ("spiky sea urchin" shape), and release their granule contents:
- ADP - recruits and activates more platelets (positive feedback)
- Thromboxane A2 - potent platelet activator and vasoconstrictor
Step 3 - Platelet Aggregation
ADP causes conformational change in glycoprotein IIb/IIIa (GpIIb-IIIa), increasing its affinity for fibrinogen. Fibrinogen bridges GpIIb-IIIa receptors on adjacent platelets, cross-linking them into a loose aggregate - the primary hemostatic plug.
Clinical correlations:
- Deficiency of vWF → von Willebrand disease (bleeding disorder)
- Deficiency of GpIb → Bernard-Soulier syndrome
- Deficiency of GpIIb-IIIa → Glanzmann thrombasthenia
Ganong emphasizes that serotonin released from activated platelets reinforces vasoconstriction at this stage, tightening the vessel around the plug.
Platelet adhesion and aggregation diagram:
4. Blood Coagulation (Secondary Hemostasis)
(Guyton & Hall + Ganong)
The primary platelet plug is weak and temporary. It must be reinforced by a fibrin meshwork through the coagulation cascade.
General Mechanism
Each reaction in the cascade involves:
- An enzyme (activated coagulation factor)
- A substrate (inactive proenzyme)
- A cofactor (reaction accelerator)
These complexes assemble on the negatively charged phospholipid surface of activated platelets, and most require Ca²⁺ for function.
Conversion of Prothrombin to Thrombin
Prothrombin activator (Factor Xa + Va + Ca²⁺ + phospholipid) cleaves prothrombin (Factor II) into thrombin. Prothrombin is a plasma protein formed in the liver; vitamin K is required for its synthesis.
Conversion of Fibrinogen to Fibrin
Thrombin cleaves fibrinogen (a soluble plasma protein) into fibrin monomers, which spontaneously polymerize into a loose fibrin mesh. Factor XIIIa (also activated by thrombin) then creates covalent cross-links between fibrin strands, making a tough, stable clot.
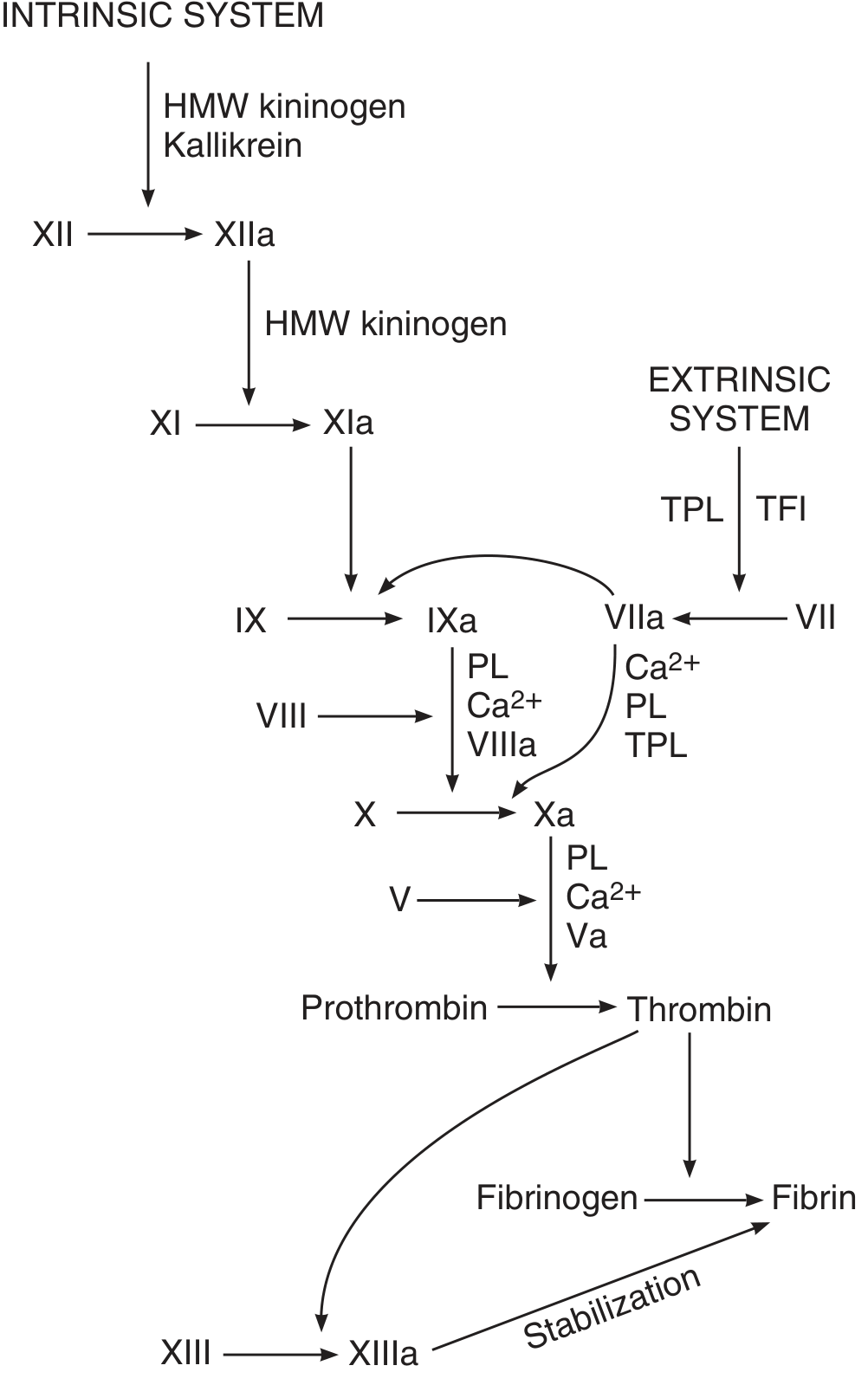
5. Initiation of Coagulation: Extrinsic vs Intrinsic Pathways
Both pathways converge on the activation of Factor X, leading to prothrombin activator formation.
Extrinsic Pathway (faster, ~15 seconds)
Triggered by tissue trauma:
- Damaged tissue releases tissue thromboplastin (tissue factor, Factor III)
- Tissue factor + Factor VII + Ca²⁺ → Factor VIIa complex
- This directly activates Factor X
Intrinsic Pathway (slower, 1-6 minutes)
Triggered by blood trauma or contact with collagen:
- Factor XII contacts collagen or damaged vessel → Factor XIIa (aided by HMW kininogen and kallikrein)
- XIIa → Factor XIa
- XIa → Factor IXa
- IXa + Factor VIIIa + PL + Ca²⁺ → activates Factor X
Ganong notes that in vivo, the extrinsic pathway is the primary initiator, while the intrinsic pathway amplifies and sustains coagulation. Factor XII deficiency causes no clinical bleeding.
The full coagulation cascade:
6. Clot Stabilization and Retraction
(Guyton & Hall)
- Clot retraction begins within minutes of clot formation and is largely complete within 20-60 minutes
- Platelets attached to fibrin activate thrombostenin, actin, and myosin - these contractile proteins pull fibrin fibers together, compressing the clot
- This is accelerated by thrombin and Ca²⁺ released from platelet organelles
- As the clot retracts, the edges of the broken vessel are pulled together
- Serum (plasma without clotting factors) is expressed from the clot during retraction
Thrombocytopenia (low platelet count) impairs clot retraction and is a clinical sign of platelet deficiency.
7. Positive Feedback of Clot Formation
(Guyton & Hall)
Once enough thrombin is formed, it initiates a positive feedback loop:
- Thrombin acts proteolytically on prothrombin → more thrombin
- Thrombin activates Factors VIII, IX, X, XI, XII and promotes platelet aggregation
- The clot grows until blood leakage ceases
8. Anticoagulant Mechanisms (Factors That Limit Clotting)
Both books describe the critical checks that prevent unwanted clot extension:
Guyton & Hall
A. Endothelial surface factors:
- Smooth endothelial surface prevents contact activation of the intrinsic pathway
- Glycocalyx (mucopolysaccharide layer) repels platelets and clotting factors
- Thrombomodulin (membrane protein) binds and sequesters thrombin; the thrombomodulin-thrombin complex activates Protein C, which inactivates Factors Va and VIIIa
- Prostacyclin (PGI2) and nitric oxide (NO) - released by intact endothelium - inhibit platelet aggregation and are vasodilators
B. Fibrin and Antithrombin III:
- 85-90% of thrombin formed is adsorbed onto fibrin fibers, preventing spread
- Remaining thrombin is inactivated by Antithrombin III (within 12-20 minutes)
- Heparin greatly accelerates Antithrombin III activity (heparin-cofactor mechanism)
Ganong
- Thromboxane A2 vs Prostacyclin balance: TxA2 promotes platelet aggregation at the injury site; prostacyclin keeps the vessel lumen clear
- Antithrombin III inhibits active forms of Factors IX, X, XI, and XII
- Protein C pathway: Protein C (activated by thrombomodulin-thrombin complex) inactivates Factors Va and VIIIa. Congenital absence of Protein C leads to fatal uncontrolled intravascular coagulation in infancy. Resistance to activated Protein C (due to a point mutation in Factor V - "Factor V Leiden") is a common cause of thrombosis.
- Tissue Factor Pathway Inhibitor (TFPI) directly inhibits the tissue factor-VIIa complex
9. Fibrinolysis (Clot Dissolution)
(Guyton & Hall + Ganong)
Clots are temporary - once the vessel heals, the clot must dissolve:
- Plasminogen (inactive precursor) is incorporated into the forming clot
- Tissue plasminogen activator (t-PA), released from endothelial cells, cleaves plasminogen → plasmin
- Plasmin digests fibrin and fibrinogen, dissolving the clot
- Urokinase also activates plasminogen
- α2-antiplasmin inactivates free plasmin in circulation, limiting fibrinolysis to the clot site
- Plasminogen activator inhibitor (PAI) released from endothelium limits t-PA activity
Fibrin degradation products (FDPs) and D-dimers appear in the blood as the clot dissolves - clinically used as markers of thrombotic states (e.g., DVT, PE, DIC).
10. Fibrous Organization of the Blood Clot
(Guyton & Hall)
When a clot forms in a location with little to no blood flow:
- Fibroblasts invade the clot within hours and days
- A fibrous connective tissue grows into the clot
- Eventually the clot is organized into fibrous tissue, permanently sealing the vessel
11. Abnormalities of Hemostasis and Bleeding Disorders
Both books cover clinical consequences of hemostatic failure:
Vitamin K Deficiency
(Guyton & Hall)
Vitamin K is required for the liver enzyme that adds carboxyl groups to Factors II (prothrombin), VII, IX, X, and Protein C. Without this modification, these factors cannot bind Ca²⁺ and are non-functional.
- Causes: fat malabsorption, bile duct obstruction, liver disease, neonates (before gut bacteria colonize)
- Warfarin works by blocking vitamin K epoxide reductase (VKORC1)
- Treatment: IV vitamin K 4-8 hours preoperatively restores clotting
Hemophilia
(Guyton & Hall + Ganong)
- Hemophilia A (85% of cases): Factor VIII deficiency; X-linked recessive; affects ~1/10,000 males
- Hemophilia B (15%): Factor IX deficiency
- Treatment: Factor VIII concentrates (plasma-derived or recombinant DNA-produced)
Thrombocytopenia
(Guyton & Hall)
- Platelet count <50,000/µL → spontaneous bleeding into tissues
- Platelet count <20,000/µL → potentially fatal bleeding
- Characterized by petechiae (pinpoint skin hemorrhages)
von Willebrand Disease
(Ganong)
- vWF deficiency reduces platelet adhesion AND lowers plasma Factor VIII levels
- Can be congenital or acquired
- ADAMTS13 (plasma metalloprotease) cleaves large vWF multimers; deficiency in high-shear areas leads to vWF accumulation
- Desmopressin stimulates Factor VIII production and is used in mild vWD
Thrombosis
(Ganong)
- Formation of clots inside blood vessels
- Promoted by: sluggish blood flow, damaged endothelium (atherosclerotic plaques), endocardial damage
- Pulmonary embolism results from leg vein thrombi traveling to the lungs
Disseminated Intravascular Coagulation (DIC)
(Ganong)
- Complication of septicemia and extensive tissue injury
- Fibrin deposited throughout small and medium vessels
- Consumption of platelets and clotting factors causes simultaneous bleeding
- Caused by excess thrombin generation (increased tissue thromboplastin activity without adequate TFI)
Summary Table: Key Hemostatic Steps
| Step | Event | Key Players |
|---|---|---|
| 1 | Vascular spasm | TxA2, serotonin, myogenic reflex |
| 2 | Platelet adhesion | vWF, GpIb, collagen |
| 3 | Platelet activation | ADP, TxA2, shape change |
| 4 | Primary plug formation | GpIIb-IIIa, fibrinogen |
| 5 | Coagulation cascade | Extrinsic (TF-VII) + intrinsic (XII→XI→IX→VIII→X) |
| 6 | Thrombin formation | Xa + Va + PL + Ca²⁺ → thrombin |
| 7 | Fibrin clot | Thrombin → fibrinogen → fibrin; XIIIa cross-links |
| 8 | Clot stabilization | Platelet retraction, Factor XIIIa |
| 9 | Anticoagulation | Antithrombin III, Protein C, PGI2, NO, thrombomodulin |
| 10 | Fibrinolysis | t-PA → plasmin → fibrin degradation |
| 11 | Vessel repair | Fibroblast ingrowth, fibrous organization |
Sources: Guyton and Hall Textbook of Medical Physiology (9780443111013), Chapter 37 - Hemostasis and Blood Coagulation; Ganong's Review of Medical Physiology, 26th Ed (9781260122404), Chapter 31 - Blood
This is a shared conversation. Sign in to Orris to start your own chat.