Tell me everything about how to check anemia in a patient what will be the most important and tough questions related to this can be asked by the examiner and everything related to anemia as a medical ug student according to the standard book of medicine
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Searching the Web
important examiner viva questions anemia MBBS undergraduate medicine clinical examination
I now have comprehensive data from Harrison's, Goldman-Cecil, and Robbins. Let me compile the full, detailed answer.
Complete Guide to Anemia for MBBS Undergraduates
Sources: Harrison's Principles of Internal Medicine 22E, Goldman-Cecil Medicine, Robbins & Kumar Pathologic Basis of Disease, Robbins Basic Pathology
1. DEFINITION
Anemia is defined as a reduction in the red blood cell mass below normal for a given age/sex/altitude. In practice it is measured by:
- Hemoglobin (Hb) concentration
- Hematocrit (Hct / PCV)
- Red blood cell count
WHO cut-offs (Hb g/dL):
| Group | Anemia threshold |
|---|---|
| Adult males | < 13.0 |
| Adult females (non-pregnant) | < 12.0 |
| Pregnant females | < 11.0 |
| Children 6 months - 6 years | < 11.0 |
| Children 6 - 14 years | < 12.0 |
Important caveat: Hb is a concentration - plasma volume changes (e.g. pregnancy in 2nd trimester, dehydration) can artificially lower or raise it without changing total red cell mass. - Goldman-Cecil Medicine
2. PATHOPHYSIOLOGY / ERYTHROPOIESIS OVERVIEW
- Kidneys sense hypoxia (via HIF pathway) and produce erythropoietin (EPO) - normal plasma EPO: 10-25 U/L
- When Hb falls below 100-120 g/L (10-12 g/dL), EPO rises proportionally to severity of anemia
- EPO binds receptors on erythroid progenitors in marrow, inducing proliferation and maturation
- With EPO stimulation, red cell production can increase 4-5 fold within 1-2 weeks - but only if iron, B12, and folate are adequate
- Three requirements for normal erythropoiesis: (1) Normal renal EPO production, (2) Functioning erythroid marrow, (3) Adequate substrates (iron, B12, folate) - Harrison's 22E
3. CLASSIFICATION
A. By MCV (Morphological - most practical clinically)
| Type | MCV | Causes |
|---|---|---|
| Microcytic | < 80 fL | Iron deficiency, Thalassemia, Anemia of chronic disease/inflammation, Sideroblastic anemia (mnemonic: TAILS - Thalassemia, Anemia of chronic disease, Iron deficiency, Lead poisoning, Sideroblastic) |
| Normocytic | 80-100 fL | Aplastic anemia, Renal disease, Endocrinopathies, Marrow invasion, Myeloma, Pure red cell aplasia, Early iron deficiency, Acute blood loss |
| Macrocytic | > 100 fL | Oval macrocytes: B12 deficiency, Folate deficiency, Drugs (methotrexate, hydroxyurea), Myelodysplasia; Round macrocytes: Alcohol, Liver disease, Hypothyroidism, Reticulocytosis |
B. By Mechanism (Pathophysiological - examiner favorite)
| Category | Reticulocyte count | Examples |
|---|---|---|
| Underproduction (hypoproliferative) | Low | Iron deficiency, B12/folate deficiency, Aplastic anemia, Anemia of chronic disease, Renal failure, Endocrinopathy |
| Increased destruction/loss (hyperproliferative) | High | Hemolytic anemias (hereditary spherocytosis, G6PD, autoimmune), Blood loss |
| Ineffective erythropoiesis | Normal/Low (cells destroyed in marrow) | Megaloblastic anemia, Thalassemia, Myelodysplasia |
4. HISTORY TAKING IN ANEMIA (Goldman-Cecil Table 144-4)
A complete, structured history is the FIRST step. Ask:
General
- Duration: Lifelong? Chronic? Intermittent? Acute? (lifelong = likely inherited)
- Previous blood transfusions (suggests longstanding severe anemia)
- Family history of anemia, jaundice, gallstones, splenectomy
- Blood donations
Clues to Iron Deficiency / Blood Loss
- Change in bowel habits, melena (black tarry stools = upper GI bleed), hematochezia (fresh blood = lower GI bleed), diarrhea
- Hematemesis
- Menstrual history - duration of periods, number of pads/tampons per day, passage of clots
- Pica (ice = pagophagia, clay = geophagia, flour) - craving non-food substances = pathognomonic of iron deficiency
- Restless legs syndrome (RLS - strong association with iron deficiency)
- Previous GI surgeries (gastrectomy - impairs iron absorption and intrinsic factor)
- Drug history: NSAIDs (GI bleeding), PPIs, cholestyramine
Clues to Hemolysis
- Jaundice - episodic or sustained; family history of jaundice
- Dark urine - hemoglobinuria or bilirubinuria (suggests intravascular hemolysis)
- Gallstones or cholecystectomy in a young person (pigment stones from chronic hemolysis)
- Ethnic origin: thalassemia (Mediterranean, South/SE Asian), sickle cell (African, Middle Eastern), G6PD (African, Mediterranean, SE Asian)
- Exposure to oxidant drugs/fava beans (G6PD deficiency)
- Infections (Mycoplasma - cold agglutinins; Malaria)
Clues to Megaloblastic / B12 Deficiency
- Strict vegetarian or vegan diet (B12 is only in animal products)
- Previous gastric surgery, pernicious anaemia
- Neurological symptoms: tingling/numbness in hands and feet, weakness, unsteady gait (subacute combined degeneration of spinal cord - SACD)
- History of inflammatory bowel disease (Crohn's - affects terminal ileum, site of B12 absorption)
- Burning tongue, early greying of hair
Clues to Aplastic Anemia / Bone Marrow Failure
- Recurrent infections, oral candidiasis (neutropenia)
- Easy bruising, petechiae, spontaneous bleeding (thrombocytopenia)
- Drug exposure: chloramphenicol, carbamazepine, sulfonamides, gold, NSAIDs, benzene
- Radiation exposure
- Recent viral illness (EBV, hepatitis)
- Paroxysmal nocturnal hemoglobinuria (PNH) features - nocturnal dark urine
Clues to Systemic Disease
- Symptoms of rheumatoid arthritis, SLE, IBD (anemia of inflammation)
- Renal disease - chronic kidney disease (reduced EPO)
- Weight loss, night sweats, lymphadenopathy (malignancy)
- Hypothyroid symptoms (cold intolerance, constipation, weight gain)
- Alcohol intake (macrocytosis, folate deficiency, liver disease)
5. PHYSICAL EXAMINATION IN ANEMIA (Goldman-Cecil Table 144-5)
General Signs of Anemia (Pallor)
Look for pallor at 5 standard sites:
- Conjunctival pallor (lower palpebral conjunctiva) - most reliable
- Palmar pallor (compare with your own hand)
- Nail bed pallor
- Buccal mucosa pallor
- Tongue pallor
Vital Signs
- Tachycardia - compensation
- Flow murmur (systolic, ejection type) - hyperdynamic circulation
- Orthostatic hypotension (acute blood loss)
Specific Signs by Type
| Sign | Suggests |
|---|---|
| Koilonychia (spoon-shaped nails) | Iron deficiency |
| Angular cheilitis / glossitis | Iron deficiency, B12/folate deficiency |
| Atrophic glossitis (smooth, beefy-red tongue) | Iron deficiency, B12 deficiency |
| Jaundice + pallor | Hemolytic anemia |
| Splenomegaly | Hemolysis, thalassemia, hereditary spherocytosis, myeloproliferative disease |
| Frontal bossing + prominent malar eminences | Thalassemia major (extramedullary hematopoiesis causes marrow expansion) |
| Leg ulcers | Sickle cell anemia |
| Peripheral neuropathy, posterior column signs (loss of vibration, proprioception) | B12 deficiency (SACD) |
| Petechiae / purpura | Aplastic anemia (thrombocytopenia) |
| Lymphadenopathy | Lymphoma, leukemia, infections |
| Hepatosplenomegaly | Hematological malignancy, thalassemia |
| Rectal exam - heme-positive stool | Blood loss / GI cause |
| Telangiectasias | Hereditary hemorrhagic telangiectasia |
| Bone tenderness (sternal, tibial) | Leukemia / marrow infiltration |
Signs Affecting Interpretation
- Edema/anasarca - dilutes Hb concentration, making anemia appear worse
- Dehydration / orthostatic hypotension - concentrates Hb, masking anemia
6. LABORATORY EVALUATION (Step-by-Step Approach)
Step 1: CBC with Indices
- Hemoglobin / Hematocrit
- MCV (mean corpuscular volume) - categorizes morphology
- MCH (mean corpuscular hemoglobin)
- MCHC (mean corpuscular hemoglobin concentration) - low in iron deficiency, normal/high in spherocytosis
- RDW (red cell distribution width) - elevated in iron deficiency; normal in thalassemia trait (useful distinguishing feature!)
- WBC and platelet count - pancytopenia suggests aplastic anemia or marrow infiltration
Step 2: Reticulocyte Count
- Always interpret as reticulocyte production index (RPI) or absolute reticulocyte count
- Formula for corrected reticulocyte count (CRC):
(Observed Hct / Normal Hct) x Reticulocyte % - RPI = CRC / maturation correction (1 if Hct >35%; 1.5 if 25-35%; 2 if 15-25%; 2.5 if <15%)
- RPI > 2-3: Adequate marrow response = blood loss or hemolysis
- RPI < 2: Inadequate response = hypoproliferative or ineffective erythropoiesis
Step 3: Peripheral Blood Smear - "READ THE SMEAR!"
This is the single most important test an examiner will quiz you on.
| Smear Finding | Diagnosis |
|---|---|
| Hypochromic microcytes + pencil cells (cigar cells) | Iron deficiency |
| Macro-ovalocytes + hypersegmented neutrophils (≥5 lobes in >5% or any cell with ≥6 lobes) | Megaloblastic anemia |
| Spherocytes (small, dense, no central pallor) | Hereditary spherocytosis, autoimmune hemolytic anemia |
| Target cells (codocytes) | Thalassemia, hemoglobinopathy, liver disease, post-splenectomy |
| Sickle cells | Sickle cell anemia |
| Schistocytes (red cell fragments) | Microangiopathic hemolytic anemia (TTP, HUS, DIC), prosthetic valve hemolysis |
| Tear-drop cells (dacrocytes) | Myelofibrosis, marrow infiltration |
| Basophilic stippling | Lead poisoning, thalassemia, sideroblastic anemia |
| Heinz bodies (require special stain) | G6PD deficiency, unstable hemoglobins |
| Bite cells (blister cells) | G6PD deficiency |
| Rouleaux formation | Multiple myeloma, Waldenstrom's macroglobulinemia |
| Polychromasia (bluish-grey cells = young RBCs with RNA) | Reticulocytosis (hemolysis, blood loss) |
| Nucleated RBCs | Severe hemolysis, asplenia, marrow infiltration, severe anemia |
Iron deficiency peripheral smear (Robbins):
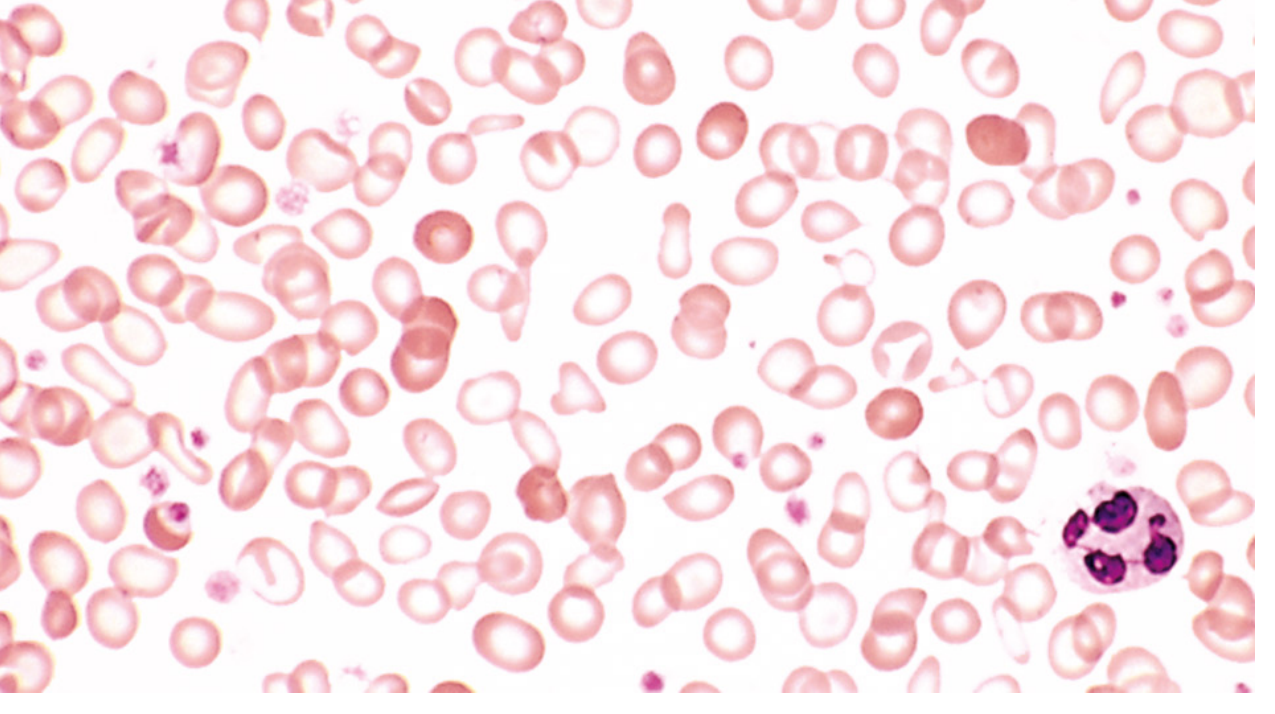
Note the hypochromic microcytic RBCs with enlarged central pallor, and pencil cells. The darker cells are from recent transfusion. - Robbins Pathology
7. SPECIFIC INVESTIGATIONS BY TYPE
Iron Studies
| Parameter | Iron Deficiency | Anemia of Chronic Disease | Thalassemia |
|---|---|---|---|
| Serum iron | ↓ | ↓ | Normal / ↑ |
| TIBC (transferrin) | ↑ | ↓ / Normal | Normal |
| Transferrin saturation | ↓ (<15%) | ↓ | Normal / ↑ |
| Serum ferritin | ↓ (< 12 μg/L) | ↑ / Normal | Normal / ↑ |
| Serum hepcidin | ↓ | ↑ | ↓ |
| Bone marrow iron (Prussian blue) | Absent | Normal/Increased | Normal |
Key concept: Ferritin is an acute phase reactant - it can be falsely normal or elevated in iron deficiency WITH concurrent inflammation. In this case, serum ferritin < 30-50 μg/L should raise suspicion for iron deficiency. - Harrison's 22E
B12 and Folate Studies
- Serum B12 (< 200 pg/mL = deficient; 200-300 = borderline)
- Serum/RBC folate
- Serum homocysteine: elevated in BOTH B12 and folate deficiency
- Serum methylmalonic acid (MMA): elevated ONLY in B12 deficiency (not folate) - this is the most specific test
- Anti-intrinsic factor antibodies - positive in pernicious anemia (highly specific)
- Anti-parietal cell antibodies - positive in ~90% but less specific
Hemolysis Screen
- Serum bilirubin (unconjugated ↑ in hemolysis)
- Serum LDH (↑ in hemolysis, especially intravascular)
- Serum haptoglobin (↓ in hemolysis - haptoglobin binds free Hb and is consumed)
- Urine hemoglobin / hemosiderin (intravascular hemolysis only)
- Direct Coombs test (DAT) - positive in autoimmune hemolytic anemia
- Osmotic fragility test (hereditary spherocytosis)
- G6PD assay
- Hemoglobin electrophoresis (thalassemia, sickle cell)
Bone Marrow Examination (when to do it)
Indicated in: Aplastic anemia, marrow infiltration, suspected hematological malignancy, unexplained pancytopenia, refractory anemia
8. INDIVIDUAL TYPES OF ANEMIA
A. Iron Deficiency Anemia (IDA)
Most common nutritional disorder in the world. (Robbins)
Stages of iron deficiency:
- Pre-latent: Iron stores depleted (↓ ferritin), Hb normal
- Latent: Iron transport deficient (↓ serum iron, ↑ TIBC, ↓ transferrin sat), Hb normal
- Iron deficiency anemia: ↓ Hb, microcytic hypochromic RBCs
Causes:
- Increased demand: Pregnancy, infancy, rapid growth
- Decreased intake: Poor diet, malabsorption (celiac disease, post-gastrectomy)
- Increased loss: Menorrhagia (most common in women of childbearing age), GI bleeding (most common in adult men and postmenopausal women)
Clinical features:
- General anemia symptoms + koilonychia, angular cheilitis, glossitis, pica, restless legs
- Plummer-Vinson syndrome (rare triad): Dysphagia (post-cricoid web) + microcytic hypochromic anemia + atrophic glossitis
- Pica: depletion of iron-containing enzymes in CNS cells. Types: pagophagia (ice), geophagia (clay/dirt), amylophagia (starch/flour)
Lab: ↓Hb, ↓MCV, ↓MCH, ↓MCHC, ↑RDW, ↓serum iron, ↑TIBC, ↓transferrin saturation (<15%), ↓ferritin, ↓hepcidin
Treatment: Oral ferrous sulfate 325 mg TDS; expect reticulocyte peak at 5-10 days, Hb increases ~1 g/dL/week. Continue 3-6 months after normalization to replenish stores.
B. Megaloblastic Anemia (B12 and Folate Deficiency)
Mechanism: Deficiency of B12 or folate → inadequate thymidine synthesis → defective DNA replication → nuclear-cytoplasmic dyssynchrony → enlarged abnormal precursors (megaloblasts) → ineffective erythropoiesis → macrocytic anemia and (usually) pancytopenia - Robbins Basic Pathology
Key distinguishing features B12 vs Folate:
| Feature | B12 Deficiency | Folate Deficiency |
|---|---|---|
| Neurological damage | YES (SACD - posterior + lateral columns) | NO |
| Serum homocysteine | ↑ | ↑ |
| Serum MMA | ↑ | Normal |
| Common cause | Pernicious anemia, veganism, gastric surgery, Crohn's (terminal ileum) | Poor diet, alcoholism, pregnancy, phenytoin, methotrexate |
| Response to folate | YES (hematologic) - but will NOT fix neuro damage, may worsen it | YES |
Critically important: Always exclude B12 deficiency BEFORE giving folate alone, because folate can correct the blood picture but will NOT prevent (and may exacerbate) the neurological complications of B12 deficiency. - Robbins
Smear: Macro-ovalocytes + hypersegmented neutrophils (hallmark)
Bone marrow: Hypercellular with megaloblasts
Causes of B12 deficiency:
- Pernicious anemia (autoimmune destruction of parietal cells - reduced intrinsic factor)
- Gastrectomy / gastric bypass
- Strict vegetarians/vegans (no animal products in diet)
- Crohn's disease (terminal ileum - site of B12-IF complex absorption)
- Fish tapeworm (Diphyllobothrium latum) - consumes B12
C. Anemia of Chronic Disease / Inflammation
Mechanism: Chronic infection, immune disorders, or neoplasia → inflammatory cytokines (IL-6) → ↑ hepcidin production by liver → hepcidin blocks ferroportin → iron trapped in macrophages, impaired iron absorption from gut → iron unavailable for erythropoiesis + direct EPO suppression. - Robbins
Key: Iron stores are NORMAL or INCREASED but iron is locked up (functional iron deficiency).
Lab: ↓ serum iron, ↓ TIBC (key distinction from IDA!), ↑ ferritin, ↑ hepcidin, normocytic/normochromic (may be mildly microcytic)
Treatment: Treat the underlying disease. EPO agents if CKD.
D. Aplastic Anemia
Definition: Bone marrow failure resulting in hypocellular marrow and peripheral pancytopenia.
Causes:
- Idiopathic (most common - immune mediated T-cell destruction of stem cells)
- Drugs: chloramphenicol, NSAIDs, carbamazepine, gold, benzene (dose-independent idiosyncratic reactions)
- Radiation
- Viral: EBV, hepatitis (non-A, non-B, non-C), HIV, parvovirus B19
- Inherited: Fanconi's anemia (defective DNA repair), dyskeratosis congenita (telomerase defect)
- PNH (paroxysmal nocturnal hemoglobinuria)
Clinical triad: Anemia + bleeding (thrombocytopenia) + infections (neutropenia)
Diagnosis: Bone marrow biopsy showing hypocellular marrow with fat replacement
Criteria for severe aplastic anemia (Camitta criteria):
- BM cellularity < 25% AND any two of: Neutrophils < 0.5 × 10⁹/L, Platelets < 20 × 10⁹/L, Reticulocytes < 20 × 10⁹/L
Treatment: Allogeneic BMT (young patients with matched sibling donor) or immunosuppression (ATG + cyclosporine + eltrombopag)
E. Hemolytic Anemia
Classification:
- Intracorpuscular (intrinsic RBC defect) - usually hereditary
- Membrane defects: Hereditary spherocytosis (spectrin/ankyrin defect), hereditary elliptocytosis
- Enzyme defects: G6PD deficiency, Pyruvate kinase deficiency
- Hemoglobin defects: Sickle cell anemia, Thalassemia
- Extracorpuscular (extrinsic factors) - usually acquired
- Immune: Autoimmune (warm IgG or cold IgM), drug-induced
- Mechanical: Microangiopathic (TTP, HUS, DIC), prosthetic valve
- Infections: Malaria, Clostridium
- Hypersplenism
Lab markers of hemolysis:
- ↑ Unconjugated bilirubin → jaundice
- ↑ LDH
- ↓ Haptoglobin (most sensitive test)
- ↑ Reticulocytes
- Hemoglobinuria, hemosiderinuria (intravascular only)
Intravascular vs Extravascular hemolysis:
| Feature | Intravascular | Extravascular |
|---|---|---|
| Site of RBC destruction | Within blood vessels | Spleen/liver macrophages |
| Hemoglobinuria | YES | No |
| Haptoglobin | ↓↓ (more severely) | ↓ |
| Splenomegaly | Variable | YES |
| Examples | TTP, G6PD crisis, PNH, transfusion reaction | Hereditary spherocytosis, autoimmune HA |
F. Thalassemia
- α-thalassemia: Deletion of α-globin genes (chromosome 16). 4-gene deletion = Hb Bart's (hydrops fetalis). 3-gene deletion = HbH disease.
- β-thalassemia major: β⁰/β⁰ or β⁰/β⁺ mutations. Presents in first 2 years of life. Severe microcytic anemia, massive splenomegaly, frontal bossing (chipmunk/thalassemic facies), "hair on end" appearance on skull X-ray (due to marrow expansion), growth retardation.
- β-thalassemia trait: Mild microcytic anemia. RDW normal (uniform microcytosis) vs iron deficiency where RDW is elevated. Hb electrophoresis: ↑ HbA2 (>3.5%) - DIAGNOSTIC.
- Treatment: Regular transfusions + iron chelation (desferrioxamine/deferasirox), curative BMT.
G. Sickle Cell Anemia
- Substitution of glutamic acid → valine at position 6 of β-globin chain
- HbS polymerizes under low O₂, acidosis, or dehydration → sickle-shaped cells → vaso-occlusion + hemolysis
- Clinical: Painful crises (vaso-occlusive), acute chest syndrome, stroke, splenic sequestration, aplastic crisis (parvovirus B19), priapism, avascular necrosis (femoral head), leg ulcers, autosplenectomy → susceptibility to encapsulated organisms (Streptococcus pneumoniae, H. influenzae, Salmonella osteomyelitis)
- Diagnosis: Hb electrophoresis (HbSS), sickle solubility test
- Treatment: Hydroxyurea (increases HbF production), penicillin prophylaxis, vaccination, pain management, BMT (curative)
9. COMPENSATION FOR ANEMIA
Three physiologic mechanisms (Harrison's 22E):
- ↑ Cardiac output (minutes) - most immediate; limited by cardiac reserve
- ↑ 2,3-DPG (2,3-diphosphoglycerate) - shifts oxyhemoglobin dissociation curve right, delivering more O₂ to tissues at lower PO₂ (hours)
- ↑ EPO with stimulation of erythropoiesis (days-weeks)
Patients who develop anemia gradually (e.g. iron deficiency over months) can tolerate remarkably low Hb levels (even 4-5 g/dL) because all three compensation mechanisms are maximally activated. Acute anemia (e.g. trauma) is far less well-tolerated.
10. TRANSFUSION TRIGGERS
- Symptomatic anemia regardless of Hb
- Hb < 7 g/dL in stable patients (restrictive strategy is safe in most)
- Hb < 8 g/dL in cardiac disease or orthopedic surgery patients
- Active hemorrhage with hemodynamic instability
11. TOUGH EXAMINER QUESTIONS (with Model Answers)
Viva / Clinical Exam Questions
Q1. What is the single most specific test to distinguish iron deficiency anemia from anemia of chronic disease?
Bone marrow biopsy with Prussian blue stain - absent iron in IDA, normal/increased iron in ACD. In practice, serum ferritin < 12 μg/L is diagnostic of IDA; TIBC is elevated in IDA but reduced in ACD.
Q2. Why does the RDW help distinguish IDA from thalassemia trait?
In iron deficiency, RBCs are heterogeneously small (some new, some old) → high RDW (anisocytosis). In thalassemia trait, all RBCs are uniformly small → RDW is normal. Use the Mentzer index: MCV/RBC count. If >13 suggests IDA; if <13 suggests thalassemia.
Q3. A patient has macrocytic anemia. You give folic acid and the Hb improves but the patient develops worsening of neurological symptoms. Explain.
This is B12 deficiency being masked. Folate can correct the hematological abnormality in B12 deficiency but will NOT correct (and may actually unmask or worsen) the neurological damage (subacute combined degeneration of spinal cord). This is why you must always rule out B12 deficiency before treating with folate alone.
Q4. What is the reticulocyte production index and how do you calculate it? What does it tell you?
RPI = (Patient's Hct / Normal Hct) × Reticulocyte % / Maturation factor. RPI > 2-3 = bone marrow is responding appropriately → implies blood loss or hemolysis. RPI < 2 = inadequate marrow response → implies hypoproliferative or ineffective erythropoiesis.
Q5. What is hepcidin? Why is it important in anemia?
Hepcidin is a peptide hormone produced by the liver. It is the master regulator of iron homeostasis. It works by binding to and degrading ferroportin (the only known iron export protein) on enterocytes, macrophages, and hepatocytes, blocking iron release into plasma. In ACD: IL-6 → ↑ hepcidin → iron trapped in macrophages. In IDA: iron stores depleted → ↓ hepcidin → maximal iron absorption. In hereditary hemochromatosis: mutations cause ↓ hepcidin → uncontrolled iron absorption.
Q6. What is pernicious anemia? How is it diagnosed?
Pernicious anemia is an autoimmune gastritis causing destruction of gastric parietal cells → loss of intrinsic factor (IF) → inability to absorb B12 in terminal ileum → megaloblastic anemia + SACD. Diagnosed by: positive anti-intrinsic factor antibodies (specific but only 60% sensitive) and/or anti-parietal cell antibodies (~90% sensitive, less specific). Schilling test (now rarely done) confirms absorption defect corrected by IF.
Q7. Hypersegmented neutrophils are seen on smear. What is your diagnosis and what are the causes?
Hypersegmented neutrophils (≥5 lobes in >5% of neutrophils, or any cell with ≥6 lobes) = megaloblastic change. Causes: B12 deficiency, folate deficiency, hydroxyurea, methotrexate, and certain myelodysplastic syndromes.
Q8. A 25-year-old woman has Hb 9 g/dL with low MCV. Serum ferritin is 45 μg/L. Is she iron deficient?
Ferritin is an acute phase reactant. In the presence of inflammation, infection, or malignancy, ferritin can be falsely elevated, masking underlying iron deficiency. A ferritin of 45 μg/L does not exclude iron deficiency in this setting. A serum ferritin < 100 μg/L combined with transferrin saturation < 20% in a CKD patient, or a therapeutic trial of iron, may be warranted.
Q9. What is the difference between intravascular and extravascular hemolysis? Give two examples of each.
Intravascular: RBCs lysed within blood vessels → free Hb released → hemoglobinuria (dark/cola urine), ↓↓ haptoglobin. Examples: TTP/HUS (schistocytes), G6PD crisis, transfusion reactions, PNH. Extravascular: RBCs opsonized → destroyed by splenic/hepatic macrophages → no hemoglobinuria, splenomegaly. Examples: Hereditary spherocytosis, autoimmune hemolytic anemia (warm AIHA), thalassemia.
Q10. Why does sickle cell cause splenic infarction and autosplenectomy?
Sickle cells cause vaso-occlusion in the small sinusoidal vessels of the spleen. Repeated sickling episodes → progressive splenic infarction → fibrosis → shrunken, non-functional spleen (autosplenectomy) by age 5 years in homozygous HbSS. This causes hyposplenism → inability to opsonize encapsulated bacteria → susceptibility to Streptococcus pneumoniae, H. influenzae type b, Salmonella.
Q11. What is Plummer-Vinson syndrome?
A rare triad of: (1) Post-cricoid esophageal web causing dysphagia, (2) Microcytic hypochromic anemia, (3) Atrophic glossitis. Occurs in middle-aged women with iron deficiency. Associated with increased risk of post-cricoid carcinoma. Treatment: Iron supplementation + endoscopic web dilatation.
Q12. What is the "anemia of renal failure" and why does it occur?
In chronic kidney disease, damaged kidneys fail to produce adequate EPO in response to anemia → hypoproliferative normocytic normochromic anemia. Additional factors include: uremic toxin inhibition of erythropoiesis, shortened RBC survival, blood loss from hemodialysis. Treatment: Recombinant EPO (erythropoiesis-stimulating agents) with IV iron supplementation.
Q13. Patient has aplastic anemia. What are the criteria for "severe" disease and why does it matter?
Camitta criteria for severe aplastic anemia: BM cellularity < 25% AND ≥2 of: neutrophils < 0.5 × 10⁹/L, platelets < 20 × 10⁹/L, reticulocytes < 20 × 10⁹/L. It matters because severe aplastic anemia requires urgent treatment - allogeneic BMT (in young patients with HLA-matched sibling) or immunosuppression with antithymocyte globulin + cyclosporine. Very severe = neutrophils < 0.2 × 10⁹/L.
Q14. On smear you see schistocytes and low platelets. What is your diagnosis and what is the pathophysiology?
This is microangiopathic hemolytic anemia (MAHA). Most likely TTP (thrombotic thrombocytopenic purpura) or HUS. Pathophysiology: Deficiency of ADAMTS-13 (von Willebrand factor-cleaving protease) in TTP → accumulation of large vWF multimers → platelet microthrombi in small vessels → RBCs physically sheared as they pass through → schistocytes. Classic pentad of TTP: MAHA, thrombocytopenia, fever, neurological symptoms, renal failure ("FAT RN" mnemonic).
Q15. A young child presents with severe anemia requiring transfusions and has frontal bossing. What do you suspect and what will the skull X-ray show?
Thalassemia major (β-thalassemia). The severe anemia drives massive extramedullary hematopoiesis → marrow expands in the skull diploe → "hair on end" or "crew cut" appearance on skull X-ray. Frontal bossing, prominent malar eminences, and maxillary overgrowth ("chipmunk facies") result from the same expansion.
12. SUMMARY ALGORITHM FOR ANEMIA WORKUP
Anemia (low Hb)
|
├── Get CBC + Reticulocyte count + Peripheral Smear
|
├── Reticulocyte count HIGH (RPI > 2-3)
| ├── Blood loss (check for bleeding)
| └── Hemolysis → LDH, haptoglobin, bilirubin, Coombs, smear
|
└── Reticulocyte count LOW (RPI < 2)
|
├── MCV LOW (microcytic)
| └── Iron studies (serum iron, TIBC, ferritin)
| ├── IDA: ↓ iron, ↑TIBC, ↓ ferritin
| ├── ACD: ↓ iron, ↓TIBC, ↑ ferritin
| └── Thalassemia: normal iron studies → Hb electrophoresis
|
├── MCV HIGH (macrocytic)
| └── B12, folate levels → MMA, homocysteine
|
└── MCV NORMAL (normocytic) + pancytopenia
└── Bone marrow biopsy
├── Hypocellular → Aplastic anemia
├── Hypercellular + dysplastic → MDS
└── Infiltrated → Leukemia / Metastatic carcinoma
Key Textbooks Referenced:
- Harrison's Principles of Internal Medicine, 22nd Edition (2025)
- Goldman-Cecil Medicine, International Edition
- Robbins & Kumar Pathologic Basis of Disease (latest edition)
- Robbins Basic Pathology
This covers everything a UG student (MBBS) needs to know for clinical examinations, vivas, and theory papers on anemia. The toughest examiner questions are around: reticulocyte index calculation, distinguishing IDA from ACD vs thalassemia, the B12/folate neurological pitfall, hepcidin physiology, peripheral smear interpretation, and hemolysis classification.
This is a shared conversation. Sign in to Orris to start your own chat.