Pathogenesis of neurocysticercosis
Reading File
Finding Sources
Finding Sources
Searching PubMed
neurocysticercosis pathogenesis
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
I now have all the essential material. Let me compile the comprehensive answer.
Pathogenesis of Neurocysticercosis (NCC)
Neurocysticercosis is the most common parasitic disease of the human CNS worldwide and the leading cause of acquired epilepsy in endemic regions, caused by the larval stage (cysticercus) of the pork tapeworm Taenia solium. - Harrison's Principles of Internal Medicine 22E, p. 1181
1. Transmission: How Humans Become Intermediate Hosts
The key distinction is that NCC occurs when humans inadvertently become the intermediate host (rather than the definitive host), by ingesting T. solium eggs rather than cysticerci in pork:
The Taeniasis-Cysticercosis Life Cycle:
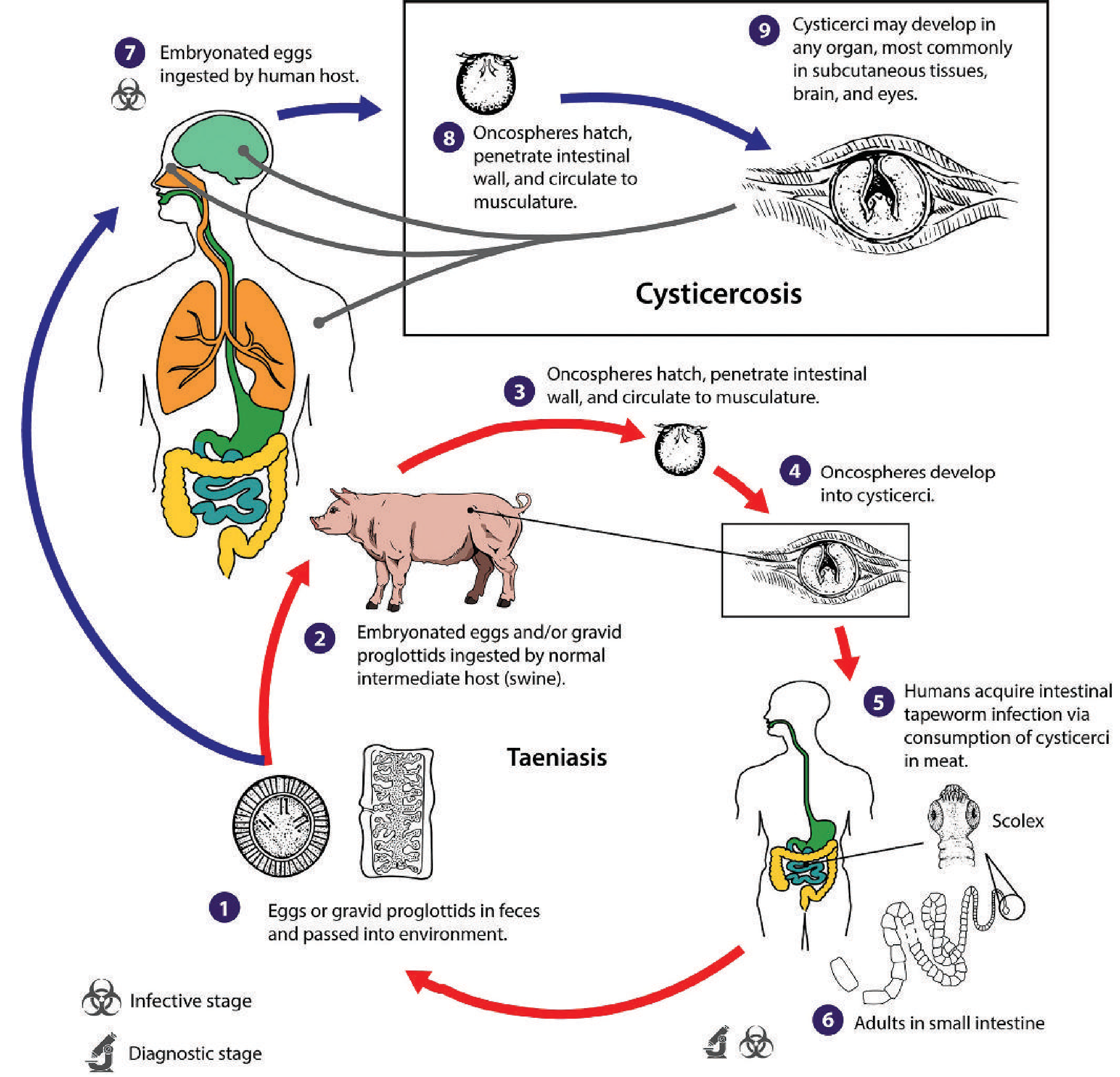
-
Definitive host route (Taeniasis): A person eats undercooked pork containing larval cysticerci → adult tapeworm develops in the small intestine → passes eggs in feces.
-
Intermediate host route (Cysticercosis / NCC): Humans ingest T. solium eggs from food, water, or fomites contaminated with infected human feces. This can occur via:
- Fecal-oral transmission from a tapeworm carrier (often a household contact or food handler)
- Auto-infection: a tapeworm carrier ingests their own eggs via reverse peristalsis or hand-to-mouth contamination
-
Harrison's: "Humans acquire cysticercosis by the ingestion of food contaminated with the eggs of the parasite Taenia solium." - p. 1181
2. Entry, Hematogenous Dissemination, and CNS Invasion
Once eggs are ingested, a precise sequence unfolds:
- Egg hatching: Gastric acid and intestinal enzymes dissolve the outer shell of the egg; the inner embryo - the oncosphere (hexacanth embryo) - is liberated in the small intestine.
- Intestinal wall penetration: The oncosphere uses its six hooks and proteolytic enzymes to penetrate the intestinal mucosa, gaining access to mesenteric venules and lymphatics.
- Hematogenous spread: Oncospheres are carried via the bloodstream to various organs - skeletal muscle, subcutaneous tissues, liver, eyes, and, most critically, the CNS.
- CNS seeding: Oncospheres preferentially lodge at the grey-white matter junction (where blood flow slows due to vessel caliber change), and in the subarachnoid space, ventricles, and occasionally spinal cord.
- Cysticercus development: Within the CNS tissue, the oncosphere transforms into a cysticercus larva over several weeks - a fluid-filled bladder worm containing a single invaginated scolex (head), surrounded by a thin outer membrane.
- Sherris & Ryan's Medical Microbiology, 8th Ed, p. 1908: "Oncospheres penetrate the intestinal wall...migrate via the bloodstream to various organs, including the skin, brain, and eyes."
3. The Four Stages of Parenchymal NCC (The Central Pathogenetic Sequence)
The central pathogenetic mechanism is the evolution of the host immune response to the lodged cysticercus. This proceeds through four well-defined stages:
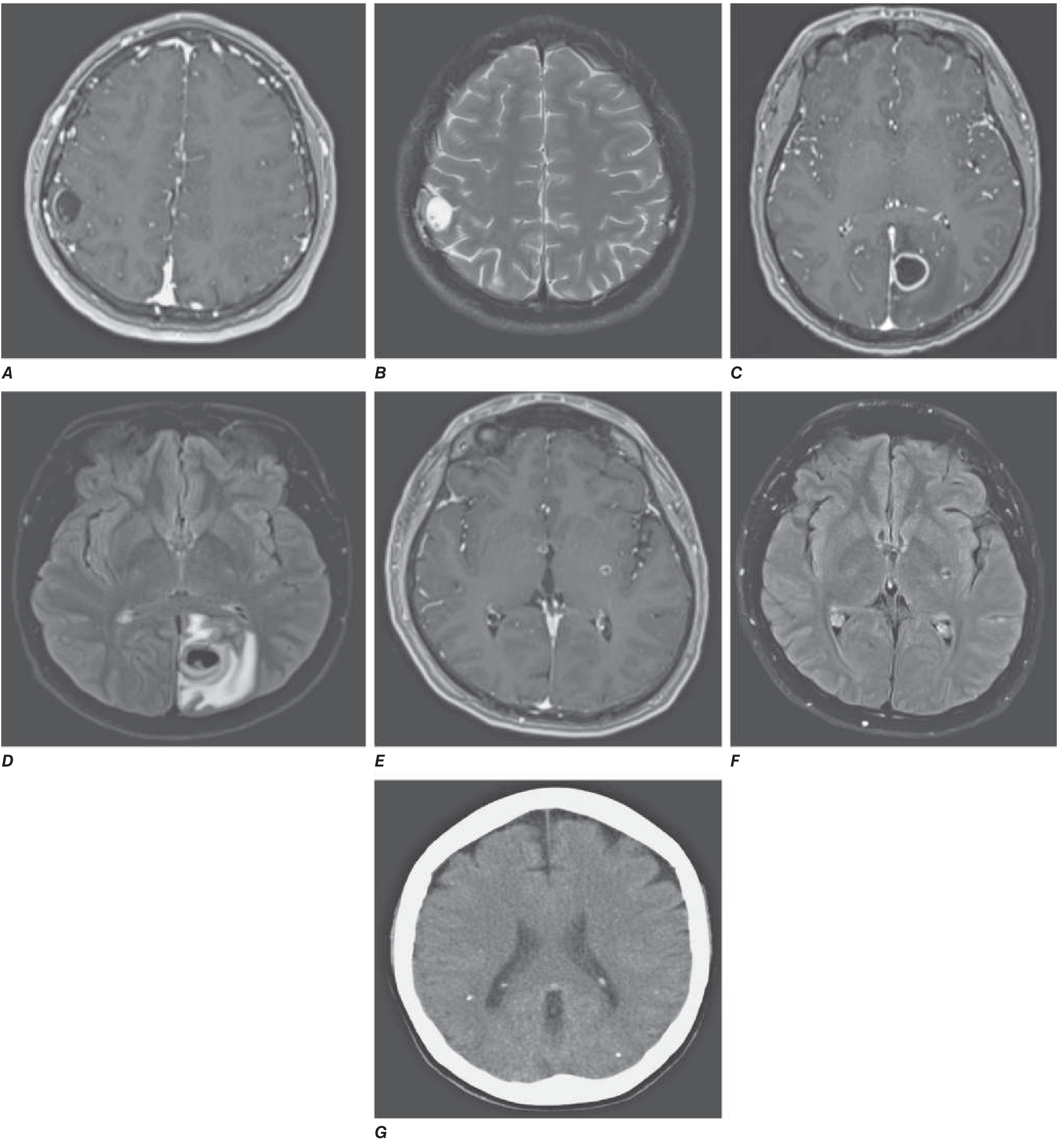
Stage 1: Vesicular (Viable Larva)
- A living larva sits within a thin-walled cyst; cyst fluid is isointense to CSF.
- The scolex is visible as a 2-5 mm eccentric nodule ("hole-with-dot" appearance on MRI).
- Little to no surrounding edema - the live parasite actively suppresses host immunity by releasing immunomodulatory molecules (parasite-derived complement inhibitors, cytokine antagonists). This is the key immune evasion phase.
- Clinically often silent for months to years.
Stage 2: Colloidal Vesicular (Early Degeneration)
- The larva begins to die; the blood-brain barrier (BBB) around the cyst breaks down.
- Release of parasite antigens triggers a vigorous host inflammatory response: mononuclear cell infiltration (lymphocytes, plasma cells, eosinophils), microglial activation.
- Imaging: ring-enhancing lesion with thick wall, marked perilesional edema, mass effect.
- Cyst fluid becomes turbid and proteinaceous (hyperintense on T1/FLAIR).
- This stage is clinically the most symptomatic - most seizures occur here due to the intense peri-lesional neuroinflammation acting as an epileptic focus.
- Harrison's: "As the cysticercal cyst degenerates, it elicits an inflammatory response that may present clinically as a seizure." - p. 1182
Stage 3: Granular Nodular (Late Degeneration)
- Cyst collapses; host macrophages and giant cells infiltrate and wall off the lesion.
- Imaging: Solid nodular enhancing lesion with thick cyst walls; surrounding edema may progress then begin to resolve.
- The lesion is partially mineralized (calcium deposition begins).
- Ongoing granulomatous inflammation - seizure risk persists.
Stage 4: Calcified Nodular (Inactive/Dead Parasite)
-
Residual calcified nodule 2-10 mm; no viable parasite remains.
-
Edema has resolved; contrast enhancement is absent (usually).
-
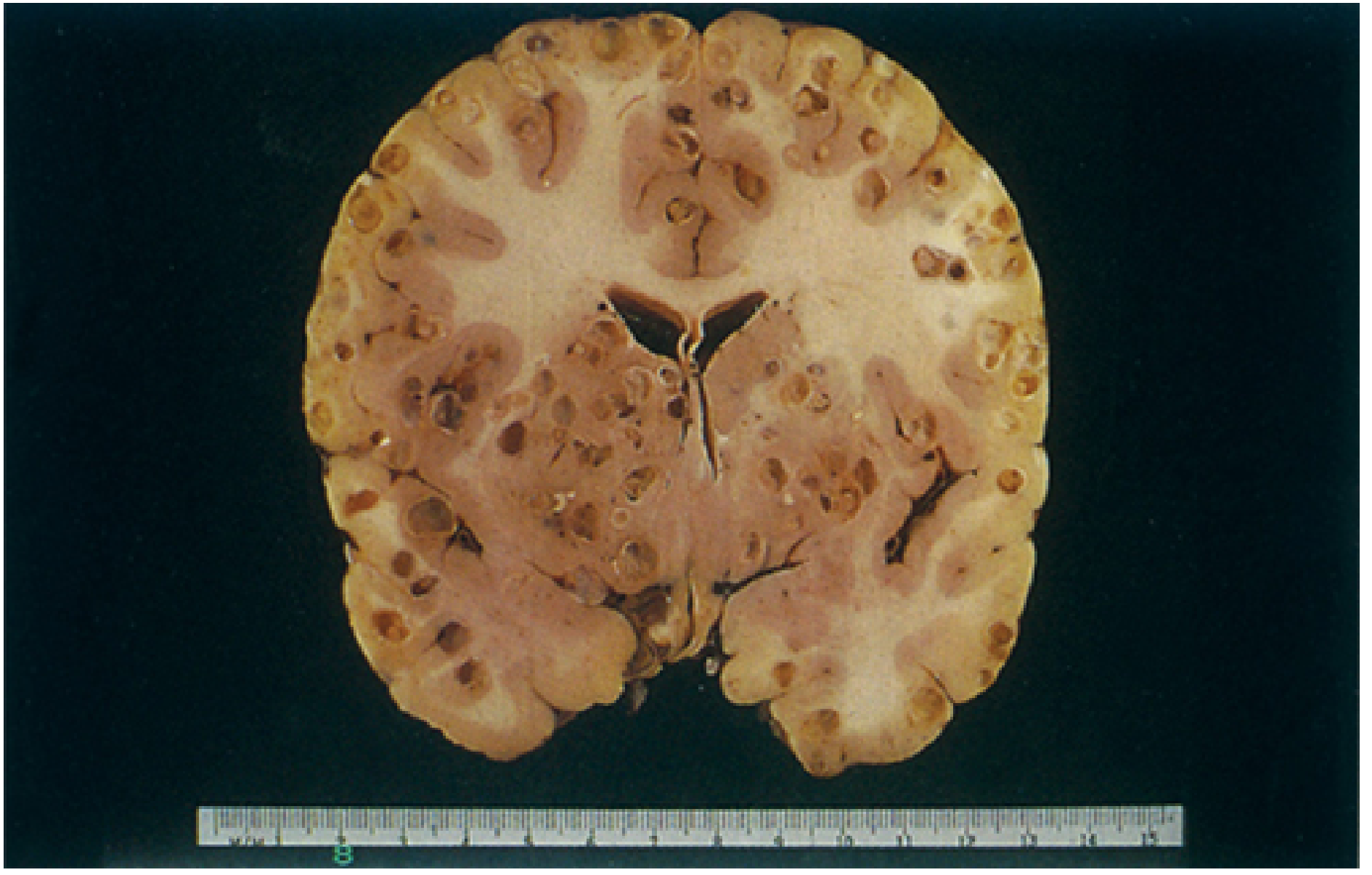
These calcified nodules are the most common finding at autopsy and on CT in endemic areas.
-
Important: even calcified lesions can intermittently provoke perilesional edema and seizures - a phenomenon thought to reflect periodic leakage of residual antigenic material or altered BBB permeability around the calcification.
-
Grainger & Allison's Diagnostic Radiology, p. 1482-1483; Harrison's 22E, p. 1183
4. Pathogenesis by Anatomical Location
The clinical syndrome in NCC is dictated by where in the CNS the cysticerci lodge, not just their viability stage:
A. Parenchymal NCC (Most Common)
- Cysts lodge at the grey-white junction, typically 2 cm or less in diameter.
- Mechanism of seizures: peri-cystic inflammation → cortical irritation → focal epilepsy (most common presentation globally).
- Focal neurologic deficits, cognitive changes, and personality disturbances occur from mass effect and gliosis.
- In 80-90% of cases, parenchymal lesions resolve spontaneously within 3-6 months. - Tintinalli's Emergency Medicine, p. 1197
B. Subarachnoid / Leptomeningeal NCC
- Cysticerci in the basal cisterns or sylvian fissures can grow unconstrained into large multiloculated "racemose" forms (clusters with grape-like septa; up to 6 cm), which carry no visible scolex.
- Cause: chronic meningitis, eosinophilic CSF pleocytosis, cranial nerve palsies.
- Racemose form incites intense local inflammation → endarteritis of leptomeningeal arteries → cerebral infarcts (stroke from infection).
- Granulomata with variable calcification form within the subarachnoid space.
C. Intraventricular NCC
- Most commonly the fourth ventricle, then third, then lateral ventricles.
- The cyst can obstruct CSF flow at the foramen of Monro, aqueduct of Sylvius, or fourth ventricle outlets → obstructive hydrocephalus with raised intracranial pressure (headache, vomiting, papilledema).
- Can cause acute, life-threatening ICP rise (Bruns syndrome: sudden severe headache/vomiting with positional change of an intraventricular cyst).
D. Spinal NCC (Rare)
-
Intramedullary or extramedullary cysts → cord compression or meningeal inflammation mimicking spinal tumor.
-
Grainger & Allison's Diagnostic Radiology, p. 1483; Sherris & Ryan's Medical Microbiology, p. 1909
5. Host Immune Response: Key Pathogenetic Driver
| Phase | Immune Event | Clinical Effect |
|---|---|---|
| Viable larva | Parasite secretes immunomodulatory factors; suppresses Th1 response; minimal inflammation | Asymptomatic |
| Early degeneration | BBB breakdown; parasite antigen release; mast cell degranulation; Th1/Th17 activation; IL-1β, TNF-α, IL-6 surge | Seizures, edema, headache |
| Late degeneration | Granuloma formation (macrophages, epithelioid cells, giant cells); fibrosis | Residual scar; ongoing seizure risk |
| Calcified stage | Low-level chronic antigen leakage; complement activation around calcification | Intermittent seizures, perilesional edema |
- The death of the larva (spontaneous or treatment-induced) is the event that maximally activates inflammation. This is why antiparasitic treatment must always be co-administered with corticosteroids to blunt the inflammatory surge. - Medical Microbiology 9e, p. 868
6. Macroscopic / Gross Pathology
Summary Flow of Pathogenesis
Ingestion of T. solium eggs (fecal-oral)
↓
Oncosphere hatches in small intestine → penetrates intestinal wall
↓
Hematogenous dissemination
↓
Oncosphere lodges in CNS (grey-white junction / subarachnoid / ventricular)
↓
Transforms into cysticercus (fluid-filled bladder worm + scolex)
↓
STAGE 1 - Vesicular: Immune evasion → silent phase
↓
STAGE 2 - Colloidal: Larval death → antigen release → BBB disruption
→ intense inflammation → perilesional edema → SEIZURES
↓
STAGE 3 - Granulonodular: Granuloma formation → cyst collapse → calcification begins
↓
STAGE 4 - Calcified: Inactive calcified nodule → residual epilepsy risk
Sources: Harrison's Principles of Internal Medicine 22E (2025), p. 1181-1183 | Grainger & Allison's Diagnostic Radiology, p. 1482-1483 | Sherris & Ryan's Medical Microbiology 8E, p. 1908-1909 | Medical Microbiology 9E, p. 868-869 | Bradley & Daroff's Neurology in Clinical Practice, p. 980 | Tintinalli's Emergency Medicine, p. 1197 | Tietz Textbook of Laboratory Medicine 7E, p. 3593
This is a shared conversation. Sign in to Orris to start your own chat.