Dead space full details for md exams 10 marks
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
In respiratory medicine exam paper
I have all the content needed from the previous research. Let me now compile this into a complete, exam-ready answer.
Dead Space — Complete Answer for MD Respiratory Medicine Exam (10 Marks)
Definition
Dead space is the volume of the respiratory system that is ventilated but does not participate in gas exchange with pulmonary capillary blood. It represents "wasted ventilation."
Dead space is a general term that refers to both the anatomic dead space of the conducting airways and a functional, or physiologic, dead space. - Costanzo Physiology 7th Ed.
Classification of Dead Space
1. Anatomic Dead Space
- The volume of gas contained in the conducting airways: nose/mouth, pharynx, larynx, trachea, bronchi, and bronchioles (up to, but not including, respiratory bronchioles and alveoli)
- These structures conduct air but have no alveolar-capillary membrane for gas exchange
- Normal value: ~150 mL in a healthy adult male (approximately 1 mL per pound of ideal body weight)
- Increases slightly with large inspirations (radial traction on bronchi expands them)
- Women have smaller anatomic dead space than men due to smaller airway caliber
- Increases slightly after age 50-60 years
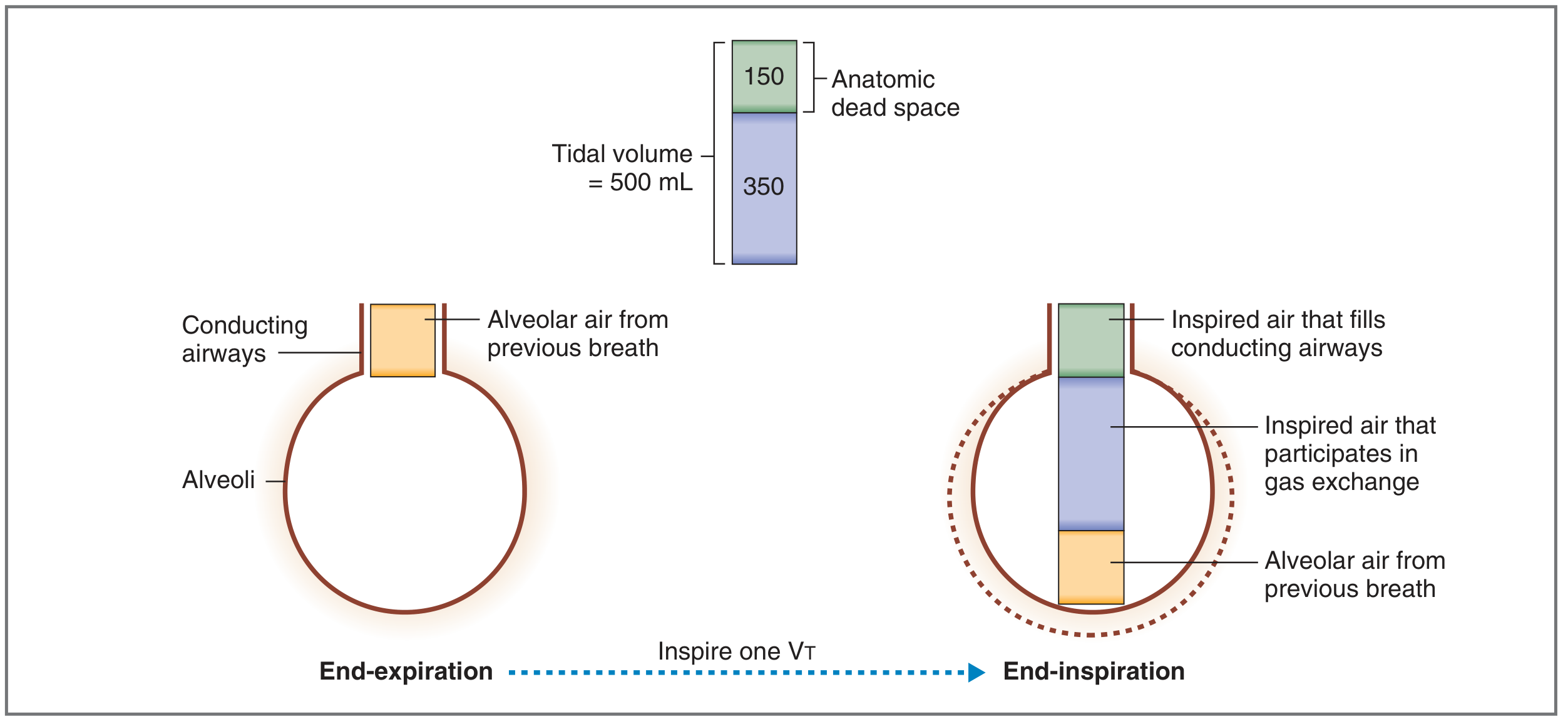
Fig: Of a 500 mL tidal volume, 150 mL fills the anatomic dead space; only 350 mL reaches the alveoli for gas exchange.
2. Alveolar Dead Space
- Volume of gas reaching alveoli that are ventilated but not perfused (or inadequately perfused) by pulmonary capillary blood
- Represents V/Q = infinity (ventilation present, perfusion absent)
- Normally negligible in healthy individuals
- Increases significantly in pathological states (e.g., pulmonary embolism, emphysema)
3. Physiologic (Total) Dead Space
Physiologic dead space = Anatomic dead space + Alveolar dead space
- This is a functional measurement based on the lung's ability to eliminate CO₂
- In healthy individuals: physiologic dead space ≅ anatomic dead space (alveolar dead space is negligible)
- In disease: physiologic dead space can exceed anatomic dead space by up to 10-fold (1-2 L)
- Normal ratio: VD/VT = ~0.3 (dead space is about one-third of tidal volume)
Measurement of Dead Space
A. Fowler's Method (Single-Breath N₂ Washout) — Measures Anatomic Dead Space
Procedure:
- Patient takes a maximal inspiration of 100% O₂ from mid-inspiration
- Then exhales steadily while expired N₂ concentration is continuously monitored
Four phases of the expired N₂ curve:
| Phase | Description |
|---|---|
| Phase I | Pure dead space gas — no N₂ (O₂ that filled conducting airways) |
| Phase II | Mixture of dead space gas and alveolar gas — N₂ rises steeply |
| Phase III | Plateau of alveolar gas — relatively constant N₂ (slight positive slope) |
| Phase IV | N₂ rises sharply = closing volume (lower zone airways close) |
- Dead space volume = volume expired from peak inspiration to the midpoint of Phase II
- The midpoint bisects Phase II such that the area "above" the curve equals the area "below" it
B. Bohr Equation — Measures Physiologic Dead Space
Based on the principle that all CO₂ in expired air comes from functioning (perfused) alveoli; dead space air contributes zero CO₂.
Derivation:
Total expired CO₂ = CO₂ from alveolar compartment + CO₂ from dead space
$$V_E \times F_{ECO_2} = V_A \times F_{ACO_2} + V_D \times F_{ICO_2}$$
Since FICO₂ ≅ 0 (room air breathing), and substituting partial pressures, and since PaCO₂ ≅ PACO₂:
$$\boxed{V_D = V_T \times \frac{P_{a}CO_2 - P_{\bar{E}}CO_2}{P_{a}CO_2}}$$
Where:
- V
D= physiologic dead space volume (mL) - V
T= tidal volume (mL) - P
aCO₂ = arterial blood PCO₂ (mm Hg) — used as a surrogate for alveolar PCO₂ - P
ECO₂ = PCO₂ of mixed expired air (mm Hg) — collected over several breaths
Example (normal):
- P
aCO₂ = 40 mmHg, PECO₂ = 28 mmHg, VT= 500 mL - V
D= 500 × (40 - 28)/40 = 500 × 0.30 = 150 mL
Extreme examples to understand the equation:
- If dead space = 0: P
ECO₂ = PaCO₂ = 40 → fraction = 0 → VD= 0 ✓ - If dead space = entire V
T: no gas exchange → PECO₂ = 0 → fraction = 1 → VD= VT✓
Key difference: Fowler's method measures anatomical dead space; the Bohr method measures physiological dead space. They yield the same result in healthy lungs. In pulmonary embolism, the Bohr method detects increased physiological dead space while Fowler's method does not. - Medical Physiology (Boron & Boulpaep)
Dead Space and Alveolar Ventilation
$$\dot{V}_A = (V_T - V_D) \times \text{Respiratory Rate}$$
Normal: (500 - 150) × 12 = 4,200 mL/min
Critical concept — Rapid shallow breathing vs. slow deep breathing:
| Parameter | Rapid Shallow | Slow Deep |
|---|---|---|
| Rate | 30/min | 10/min |
| Tidal volume | 200 mL | 600 mL |
| Minute ventilation | 6 L/min | 6 L/min |
| Alveolar ventilation | (200-150)×30 = 1,500 mL/min | (600-150)×10 = 4,500 mL/min |
Same minute ventilation, but 3x less alveolar ventilation with rapid shallow breathing — the dead space constitutes a much larger fraction of each small tidal breath.
Pathophysiology of Increased Dead Space
Causes of Increased Physiologic Dead Space
| Category | Examples |
|---|---|
| Pulmonary vascular | Pulmonary embolism (classic), decreased cardiac output, vasoconstrictors |
| Parenchymal | Emphysema (destroyed alveolar walls + capillary beds), blebs, hyperinflation |
| Mechanical | Positive pressure ventilation (compresses alveolar vessels), PEEP |
| Anatomic | Tracheostomy (bypasses upper airway dead space but adds instrument dead space) |
Mechanism of Increased Dead Space - V/Q Mismatch
- Dead space = high V/Q units (V/Q → ∞)
- Ventilated alveoli without perfusion cannot exchange O₂ or CO₂
- The alveolar PCO₂ in these units falls to inspired air levels (~0 mmHg)
- The alveolar PO₂ rises toward inspired levels (~150 mmHg)
Local Compensatory Mechanism
In the setting of increased dead space, local bronchoconstriction acts to normalize the V/Q ratio by redirecting ventilation away from underperfused regions.
Clinical Consequences of Increased Dead Space
1. Hypercapnia
- Increased dead space reduces effective CO₂ elimination
- Compensatory hyperventilation (increased minute ventilation) is the normal response
- Hypercapnia does not develop until dead space exceeds ~50% of total lung volume, because the body can increase minute ventilation to compensate
2. Hypoxemia
- Decreased PaO₂ related to dead space (V/Q > 1) generally improves with supplemental O₂ — this distinguishes it from shunt (V/Q = 0) which does not respond well to O₂
3. Increased work of breathing
- Patient must increase total minute ventilation to maintain adequate alveolar ventilation
- If ventilatory reserve is exceeded, respiratory muscle fatigue and respiratory failure ensue
VD/VT Ratio — Clinical Importance
- Normal: 0.25 - 0.35 (25-35%)
- In mechanically ventilated ICU patients: elevated VD/VT (>0.60) is associated with increased mortality in ARDS
- Used to gauge severity of V/Q mismatch in respiratory failure
- Requires arterial blood gas (for P
aCO₂) and simultaneous mixed expired gas collection
Summary Table
| Feature | Anatomic Dead Space | Alveolar Dead Space | Physiologic Dead Space |
|---|---|---|---|
| Location | Conducting airways | Non-perfused alveoli | Anatomic + Alveolar |
| Normal value | ~150 mL (~1 mL/lb IBW) | ~0 mL | ~150 mL |
| Measurement | Fowler's method (N₂ washout) | Calculated | Bohr equation (CO₂ method) |
| V/Q | No perfusion, no ventilation exchange | V/Q = ∞ | Combination |
| In disease | Minimally changed | Markedly increased | Markedly increased |
Key Exam Points to Remember
- Dead space = wasted ventilation (no gas exchange despite being ventilated)
- Physiologic DS = Anatomic + Alveolar; in health, alveolar DS ≈ 0
- Bohr equation uses arterial PCO₂ and mixed expired PCO₂ to calculate physiologic dead space
- Fowler's method (N₂ single breath) measures anatomic dead space
- Pulmonary embolism is the classic cause of increased alveolar dead space - Bohr detects it, Fowler does not
- VD/VT normal = 0.3; rises significantly in pulmonary embolism, emphysema, ARDS
- Rapid shallow breathing is highly inefficient because dead space is a larger fraction of each small tidal volume
- Hypercapnia only occurs when dead space > 50% VT because hyperventilation normally compensates
- Hypoxemia from dead space responds to O₂; hypoxemia from shunt does not
- Local bronchoconstriction is the compensatory response to reduced perfusion (redirects ventilation)
Sources: Costanzo Physiology 7th Ed. | Guyton & Hall Medical Physiology | Ganong's Review of Medical Physiology 26th Ed. | Boron & Boulpaep Medical Physiology | Fishman's Pulmonary Diseases and Disorders | Murray & Nadel's Textbook of Respiratory Medicine | Frameworks for Internal Medicine
Respiratory acidosis for md exams 10 marks answer full details
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive content from 6+ authoritative textbooks. Let me compile the full exam answer:
Respiratory Acidosis — Full Answer for MD Respiratory Medicine Exam (10 Marks)
Definition
Respiratory acidosis is a primary acid-base disorder characterized by a rise in arterial PCO₂ (hypercapnia) due to inadequate alveolar ventilation, resulting in a fall in arterial pH below 7.35.
The fundamental equation governing this process:
$$CO_2 + H_2O \underset{CA}{\rightleftharpoons} H_2CO_3 \rightleftharpoons H^+ + HCO_3^-$$
When CO₂ is retained, this reaction is driven to the right, generating excess H⁺ and lowering pH.
Henderson-Hasselbalch equation:
$$pH = 6.1 + \log\frac{[HCO_3^-]}{0.03 \times PaCO_2}$$
Kassirer-Bleich approximation (clinical bedside use):
$$[H^+] = 24 \times \frac{PaCO_2}{[HCO_3^-]}$$
Normal ABG Values (Reference)
| Parameter | Normal Range |
|---|---|
| pH | 7.35 - 7.45 |
| PaCO₂ | 35 - 45 mmHg |
| HCO₃⁻ | 22 - 26 mEq/L |
| PaO₂ | 80 - 100 mmHg |
In Respiratory Acidosis:
- pH: ↓ (< 7.35)
- PaCO₂: ↑ (primary disturbance)
- HCO₃⁻: ↑ (compensatory)
Pathophysiology
Primary Mechanism
Any condition that reduces alveolar ventilation leads to CO₂ retention. The body generates ~200 mL/min of CO₂ through oxidative metabolism. When the lung cannot excrete this load, PaCO₂ rises.
Causes reduce ventilation through four main mechanisms:
- Depression of the medullary respiratory center - reduces drive to breathe
- Impaired neuromuscular transmission - reduces mechanical ability to ventilate
- Airway obstruction - increases resistance, reduces effective ventilation
- Impaired gas exchange - CO₂ cannot exit pulmonary capillary blood into alveolar air
Sequence of Events
- Hypoventilation → CO₂ retention → ↑PaCO₂ (primary disturbance)
- ↑PCO₂ drives: CO₂ + H₂O → H⁺ + HCO₃⁻ (mass action) → pH falls, HCO₃⁻ rises slightly
- Buffering occurs exclusively in ICF - CO₂ diffuses into cells (especially RBCs), where it is converted to H⁺ + HCO₃⁻; H⁺ is buffered by intracellular proteins (hemoglobin) and organic phosphates
- No respiratory compensation exists - respiration is the cause of the disorder
- Renal compensation kicks in over 24-72 hours
There is no respiratory compensation for respiratory acidosis, since respiration is the cause of this disorder. - Costanzo Physiology 7th Ed.
Etiology / Causes
A. CNS Depression (Reduced Respiratory Drive)
- Drugs: opioids/narcotics, benzodiazepines, barbiturates, general anesthetics, alcohol
- Structural: stroke, head trauma, CNS tumors, CNS infections (encephalitis, meningitis)
- Metabolic: severe hypothyroidism (myxedema coma)
- Sleep-disordered breathing: obesity-hypoventilation syndrome (Pickwickian), primary alveolar hypoventilation (Ondine's curse), obstructive sleep apnea
B. Neuromuscular Disorders (Pump Failure)
- Lower motor neuron: poliomyelitis, amyotrophic lateral sclerosis (ALS)
- Neuromuscular junction: myasthenia gravis, botulism, organophosphate poisoning, neuromuscular blocking agents
- Muscle diseases: muscular dystrophies, polymyopathy, severe hypokalemia/hypophosphatemia
- Spinal cord injury: high cervical lesions (C3-C5 involvement paralyzes diaphragm)
- Chest wall: kyphoscoliosis, flail chest, ankylosing spondylitis
C. Airway Obstruction
- Upper airway: aspiration of foreign body, laryngospasm, angioedema, severe obstructive sleep apnea
- Lower airway: severe acute asthma, COPD exacerbation, anaphylaxis, inhalational injury
D. Parenchymal / Gas Exchange Disease
- COPD (chronic), severe pneumonia, ARDS, pulmonary edema
- Pneumothorax, massive pleural effusion, atelectasis
E. Iatrogenic / Mechanical
- Mechanical ventilation: inadequate rate/tidal volume settings, barotrauma, ET tube displacement
- Permissive hypercapnia - intentional in ARDS management (low tidal volume ventilation)
- High PEEP with reduced cardiac output (increases alveolar dead space)
- CO₂ absorption during laparoscopy
Types: Acute vs. Chronic Respiratory Acidosis
This distinction is critical clinically and in exams.
| Feature | Acute | Chronic |
|---|---|---|
| Duration | Minutes to hours | > 24-48 hours |
| Compensation | Cellular buffering only | Renal compensation fully active |
| pH | Markedly low (may be < 7.2) | Near-normal (partially compensated) |
| HCO₃⁻ rise per 10 mmHg ↑PCO₂ | 1 mEq/L | 4 mEq/L (renal) |
| H⁺ rise per mmHg ↑PCO₂ | 0.8 nEq/L | 0.3 nEq/L |
| Maximum HCO₃⁻ | - | Usually ≤ 38 mEq/L |
| Clinical urgency | Life-threatening, may need intubation | Chronic adaptation, less acute danger |
| Example | Acute severe asthma, opioid OD | COPD, obesity-hypoventilation |
Compensation: Renal Response
The kidney is the sole compensatory organ in respiratory acidosis.
Mechanism of renal compensation:
- Increased H⁺ secretion in proximal tubule and collecting duct
- Increased titratable acid excretion (H₂PO₄⁻)
- Increased NH₄⁺ synthesis and excretion
- Increased HCO₃⁻ synthesis and reabsorption (generates "new" bicarbonate)
- Chloride is excreted in exchange for retained HCO₃⁻ → hypochloremia develops
Rules of Thumb (Expected Compensation):
$$\text{Acute: } \Delta HCO_3^- = \Delta PaCO_2 \times 0.1$$
$$\text{Chronic: } \Delta HCO_3^- = \Delta PaCO_2 \times 0.4$$
Clinical application example (COPD patient):
- PaCO₂ = 70 mmHg (ΔPaCO₂ = +30 mmHg), HCO₃⁻ = 33 mEq/L (Δ = +9 mEq/L)
- Expected acute compensation: +3 mEq/L
- Expected chronic compensation: +12 mEq/L
- Actual Δ = +9 mEq/L → between acute and chronic → partially compensated chronic respiratory acidosis
If the measured HCO₃⁻ rise is greater than predicted, a concurrent metabolic alkalosis is present. If less than predicted, a concurrent metabolic acidosis is present.
Clinical Features
Neurological (CO₂ Narcosis / Hypercapnic Encephalopathy)
CO₂ is a potent cerebral vasodilator - this drives many CNS manifestations:
- Acute/rapid rise in PaCO₂: anxiety, dyspnea, confusion, psychosis, hallucinations → coma
- Chronic hypercapnia: sleep disturbances, memory loss, daytime somnolence, personality changes
- Motor: tremor, myoclonic jerks, asterixis (flapping tremor - a hallmark sign)
- Signs mimicking raised ICP: headache, papilledema, abnormal reflexes, focal weakness
- Severe acidemia (pH < 7.2): cardiac arrhythmias, reduced myocardial contractility
Cardiovascular
- Peripheral vasodilation (CO₂ is a vasodilator) → warm, flushed skin, bounding pulse
- Tachycardia (early); bradycardia and hypotension (severe)
- Pulmonary vasoconstriction (hypoxia-driven) → cor pulmonale in chronic cases
Respiratory
- Dyspnea, use of accessory muscles
- Cyanosis (from associated hypoxemia)
- Paradoxical abdominal movement (in neuromuscular failure)
Metabolic
- Hyperkalemia: H⁺ shifts into cells in exchange for K⁺ (approximately 0.5 mEq/L rise in K⁺ per 0.1 unit fall in pH)
- Hypochloremia (in chronic - Cl⁻ excreted by kidney to retain HCO₃⁻)
Diagnosis
Step 1: ABG Interpretation
- Look at pH → acidemia (< 7.35) confirms acidosis
- Look at PaCO₂ → elevated (> 45 mmHg) confirms respiratory cause
- Look at HCO₃⁻ → elevated (compensation) or very low (mixed disorder)
- Calculate expected compensation using rules of thumb
- If compensation is appropriate → simple respiratory acidosis
- If HCO₃⁻ is higher than expected → mixed respiratory acidosis + metabolic alkalosis
- If HCO₃⁻ is lower than expected → mixed respiratory acidosis + metabolic acidosis
Step 2: Identify Acute vs. Chronic
- ABG + clinical history (duration of symptoms, prior ABGs if available)
Step 3: Workup for Cause
- Chest X-ray - first-line investigation
- Pulmonary function tests (spirometry, DLCO, lung volumes) - if lung disease suspected
- Drug history - always exclude opioids, sedatives
- Hematocrit - anemia
- Neurological assessment - if CNS cause suspected (CT head, MRI spine)
- Neuromuscular assessment - NCS/EMG, acetylcholine receptor antibodies (myasthenia)
- Polysomnography - if sleep-disordered breathing suspected
Treatment
Principles
The management depends on severity, rate of onset, and underlying cause.
1. Acute Respiratory Acidosis (Life-threatening)
- Reverse the underlying cause simultaneously with restoration of ventilation
- Tracheal intubation and mechanical ventilation if indicated
- Treat bronchospasm (acute asthma): nebulized bronchodilators, IV corticosteroids, magnesium sulfate
- Reverse opioid toxicity: naloxone (IV/IM/intranasal)
- Reverse benzodiazepine toxicity: flumazenil
2. Oxygen Therapy - Special Caution in COPD
- O₂ must be titrated carefully in chronic CO₂ retainers (COPD with chronic hypercapnia)
- These patients rely on hypoxic drive; injudicious O₂ → removes hypoxic stimulus → further hypoventilation → worsening respiratory acidosis
- Target SpO₂: 88-92% in chronic hypercapnic COPD (not 94-98%)
- Non-invasive ventilation (NIV/BiPAP) is preferred over intubation in COPD exacerbation
3. Chronic Respiratory Acidosis
- Aims at improving underlying lung function
- Smoking cessation, bronchodilators, corticosteroids, diuretics (pulmonary edema), physiotherapy
- Long-term NIV (BiPAP) at home for obesity-hypoventilation syndrome, neuromuscular disease
- Treatment of sleep apnea: CPAP/BiPAP
4. Correction of PaCO₂ - Important Caveat
- Avoid aggressive rapid correction of hypercapnia in chronic respiratory acidosis
- Rapidly falling PaCO₂ → same complications as acute respiratory alkalosis: cardiac arrhythmias, seizures, reduced cerebral perfusion
- Lower PaCO₂ gradually to baseline level (not necessarily normal)
- Simultaneously provide adequate Cl⁻ and K⁺ to enable renal excretion of the retained HCO₃⁻
5. Permissive Hypercapnia (in ARDS/ICU)
- Lung-protective ventilation uses low tidal volumes (6 mL/kg IBW) to prevent barotrauma
- Allows PaCO₂ to rise (permissive hypercapnia)
- Acceptable pH target: approximately 7.20-7.25
- If concurrent metabolic acidosis, may require sodium bicarbonate infusion
- Absolute contraindication: raised intracranial pressure (CO₂-driven vasodilation worsens ICP)
Summary: Key Points Table
| Feature | Detail |
|---|---|
| Primary disturbance | ↑ PaCO₂ (hypoventilation) |
| ABG pattern | pH ↓, PaCO₂ ↑, HCO₃⁻ ↑ |
| Compensation | Renal (no respiratory compensation) |
| Acute HCO₃⁻ rise | 1 mEq/L per 10 mmHg ↑PaCO₂ |
| Chronic HCO₃⁻ rise | 4 mEq/L per 10 mmHg ↑PaCO₂ |
| Max HCO₃⁻ in compensation | ~38 mEq/L |
| Classic signs | Asterixis, papilledema, CO₂ narcosis |
| Most common chronic cause | COPD |
| Most common acute cause | Opioid overdose / acute severe asthma |
| O₂ caution | Titrate carefully in chronic CO₂ retainers (target SpO₂ 88-92%) |
| Correction caution | Never correct chronic hypercapnia rapidly |
Exam High-Yield Points
- Respiratory acidosis = hypoventilation = CO₂ retention - any cause of reduced alveolar ventilation
- No respiratory compensation - respiration IS the problem
- Acute vs. chronic distinction is the most important clinical differentiation - use HCO₃⁻ rules of thumb
- Asterixis is the hallmark neurological sign of CO₂ retention
- Papilledema can occur due to CO₂-driven cerebral vasodilation
- Hypochloremia develops in chronic cases as kidneys excrete Cl⁻ to retain HCO₃⁻
- Hyperkalemia occurs due to H⁺/K⁺ exchange across cell membranes
- COPD + O₂: target 88-92%, not 94-98% - hypoxic drive is critical
- Permissive hypercapnia in ARDS: deliberate strategy, pH > 7.20 is acceptable
- Mixed disorders: if HCO₃⁻ is too high → + metabolic alkalosis (e.g., vomiting + COPD); if too low → + metabolic acidosis (e.g., COPD + lactic acidosis from tissue hypoxia)
Sources: Brenner & Rector's The Kidney 10th Ed. | Costanzo Physiology 7th Ed. | Murray & Nadel's Textbook of Respiratory Medicine | Harrison's Principles of Internal Medicine 22nd Ed. | Miller's Anesthesia 10th Ed. | Sabiston Textbook of Surgery | Schwartz's Principles of Surgery
Remove dollar and give answer
Here is the same answer with all equation dollar signs removed and equations written in plain text:
Respiratory Acidosis — Full Answer for MD Respiratory Medicine Exam (10 Marks)
Definition
Respiratory acidosis is a primary acid-base disorder characterized by a rise in arterial PCO₂ (hypercapnia) due to inadequate alveolar ventilation, resulting in a fall in arterial pH below 7.35.
The fundamental equation governing this process:
CO₂ + H₂O ⇌ H₂CO₃ ⇌ H⁺ + HCO₃⁻
When CO₂ is retained, this reaction is driven to the right, generating excess H⁺ and lowering pH.
Henderson-Hasselbalch equation:
pH = 6.1 + log ( [HCO₃⁻] / 0.03 × PaCO₂ )
Kassirer-Bleich approximation (clinical bedside use):
[H⁺] = 24 × PaCO₂ / [HCO₃⁻]
Normal ABG Values (Reference)
| Parameter | Normal Range |
|---|---|
| pH | 7.35 - 7.45 |
| PaCO₂ | 35 - 45 mmHg |
| HCO₃⁻ | 22 - 26 mEq/L |
| PaO₂ | 80 - 100 mmHg |
In Respiratory Acidosis:
- pH: ↓ (< 7.35)
- PaCO₂: ↑ (primary disturbance)
- HCO₃⁻: ↑ (compensatory)
Pathophysiology
Primary Mechanism
Any condition that reduces alveolar ventilation leads to CO₂ retention. The body generates ~200 mL/min of CO₂ through oxidative metabolism. When the lung cannot excrete this load, PaCO₂ rises.
Causes reduce ventilation through four main mechanisms:
- Depression of the medullary respiratory center - reduces drive to breathe
- Impaired neuromuscular transmission - reduces mechanical ability to ventilate
- Airway obstruction - increases resistance, reduces effective ventilation
- Impaired gas exchange - CO₂ cannot exit pulmonary capillary blood into alveolar air
Sequence of Events
- Hypoventilation → CO₂ retention → ↑PaCO₂ (primary disturbance)
- ↑PCO₂ drives: CO₂ + H₂O → H⁺ + HCO₃⁻ (mass action) → pH falls, HCO₃⁻ rises slightly
- Buffering occurs exclusively in ICF - CO₂ diffuses into cells (especially RBCs), where it is converted to H⁺ + HCO₃⁻; H⁺ is buffered by intracellular proteins (hemoglobin) and organic phosphates
- No respiratory compensation exists - respiration is the cause of the disorder
- Renal compensation kicks in over 24-72 hours
There is no respiratory compensation for respiratory acidosis, since respiration is the cause of this disorder. - Costanzo Physiology 7th Ed.
Etiology / Causes
A. CNS Depression (Reduced Respiratory Drive)
- Drugs: opioids/narcotics, benzodiazepines, barbiturates, general anesthetics, alcohol
- Structural: stroke, head trauma, CNS tumors, CNS infections (encephalitis, meningitis)
- Metabolic: severe hypothyroidism (myxedema coma)
- Sleep-disordered breathing: obesity-hypoventilation syndrome (Pickwickian), primary alveolar hypoventilation (Ondine's curse), obstructive sleep apnea
B. Neuromuscular Disorders (Pump Failure)
- Lower motor neuron: poliomyelitis, amyotrophic lateral sclerosis (ALS)
- Neuromuscular junction: myasthenia gravis, botulism, organophosphate poisoning, neuromuscular blocking agents
- Muscle diseases: muscular dystrophies, polymyopathy, severe hypokalemia/hypophosphatemia
- Spinal cord injury: high cervical lesions (C3-C5 involvement paralyzes diaphragm)
- Chest wall: kyphoscoliosis, flail chest, ankylosing spondylitis
C. Airway Obstruction
- Upper airway: aspiration of foreign body, laryngospasm, angioedema, severe obstructive sleep apnea
- Lower airway: severe acute asthma, COPD exacerbation, anaphylaxis, inhalational injury
D. Parenchymal / Gas Exchange Disease
- COPD (chronic), severe pneumonia, ARDS, pulmonary edema
- Pneumothorax, massive pleural effusion, atelectasis
E. Iatrogenic / Mechanical
- Mechanical ventilation: inadequate rate/tidal volume settings, barotrauma, ET tube displacement
- Permissive hypercapnia - intentional in ARDS management (low tidal volume ventilation)
- High PEEP with reduced cardiac output (increases alveolar dead space)
- CO₂ absorption during laparoscopy
Types: Acute vs. Chronic Respiratory Acidosis
| Feature | Acute | Chronic |
|---|---|---|
| Duration | Minutes to hours | > 24-48 hours |
| Compensation | Cellular buffering only | Renal compensation fully active |
| pH | Markedly low (may be < 7.2) | Near-normal (partially compensated) |
| HCO₃⁻ rise per 10 mmHg ↑PCO₂ | 1 mEq/L | 4 mEq/L (renal) |
| H⁺ rise per mmHg ↑PCO₂ | 0.8 nEq/L | 0.3 nEq/L |
| Maximum HCO₃⁻ | - | Usually ≤ 38 mEq/L |
| Clinical urgency | Life-threatening, may need intubation | Chronic adaptation, less acute danger |
| Example | Acute severe asthma, opioid OD | COPD, obesity-hypoventilation |
Compensation: Renal Response
The kidney is the sole compensatory organ in respiratory acidosis.
Mechanism of renal compensation:
- Increased H⁺ secretion in proximal tubule and collecting duct
- Increased titratable acid excretion (H₂PO₄⁻)
- Increased NH₄⁺ synthesis and excretion
- Increased HCO₃⁻ synthesis and reabsorption (generates "new" bicarbonate)
- Chloride is excreted in exchange for retained HCO₃⁻ → hypochloremia develops
Rules of Thumb (Expected Compensation):
Acute: Delta HCO₃⁻ = Delta PaCO₂ × 0.1
Chronic: Delta HCO₃⁻ = Delta PaCO₂ × 0.4
Clinical application example (COPD patient):
- PaCO₂ = 70 mmHg (Delta PaCO₂ = +30 mmHg), HCO₃⁻ = 33 mEq/L (Delta = +9 mEq/L)
- Expected acute compensation: +3 mEq/L
- Expected chronic compensation: +12 mEq/L
- Actual Delta = +9 mEq/L → between acute and chronic → partially compensated chronic respiratory acidosis
If the measured HCO₃⁻ rise is greater than predicted → concurrent metabolic alkalosis is present. If less than predicted → concurrent metabolic acidosis is present.
Clinical Features
Neurological (CO₂ Narcosis / Hypercapnic Encephalopathy)
CO₂ is a potent cerebral vasodilator - this drives many CNS manifestations:
- Acute/rapid rise in PaCO₂: anxiety, dyspnea, confusion, psychosis, hallucinations → coma
- Chronic hypercapnia: sleep disturbances, memory loss, daytime somnolence, personality changes
- Motor: tremor, myoclonic jerks, asterixis (flapping tremor - a hallmark sign)
- Signs mimicking raised ICP: headache, papilledema, abnormal reflexes, focal weakness
- Severe acidemia (pH < 7.2): cardiac arrhythmias, reduced myocardial contractility
Cardiovascular
- Peripheral vasodilation → warm, flushed skin, bounding pulse
- Tachycardia (early); bradycardia and hypotension (severe)
- Pulmonary vasoconstriction (hypoxia-driven) → cor pulmonale in chronic cases
Respiratory
- Dyspnea, use of accessory muscles
- Cyanosis (from associated hypoxemia)
- Paradoxical abdominal movement (in neuromuscular failure)
Metabolic / Electrolytes
- Hyperkalemia: H⁺ shifts into cells in exchange for K⁺ (approximately 0.5 mEq/L rise in K⁺ per 0.1 unit fall in pH)
- Hypochloremia: in chronic - Cl⁻ excreted by kidney to retain HCO₃⁻
Diagnosis
Step 1: ABG Interpretation
- pH < 7.35 → acidemia confirmed
- PaCO₂ > 45 mmHg → respiratory origin confirmed
- HCO₃⁻ elevated → compensation (or mixed disorder)
- Calculate expected compensation using rules of thumb
- Appropriate compensation → simple respiratory acidosis
- HCO₃⁻ higher than expected → mixed respiratory acidosis + metabolic alkalosis
- HCO₃⁻ lower than expected → mixed respiratory acidosis + metabolic acidosis
Step 2: Identify Acute vs. Chronic
- ABG + clinical history (duration of symptoms, prior ABGs)
Step 3: Workup for Cause
- Chest X-ray - first-line investigation
- Pulmonary function tests (spirometry, DLCO, lung volumes)
- Drug history - always exclude opioids, sedatives
- Hematocrit - anemia
- Neurological assessment - CT head, MRI spine if CNS cause suspected
- Neuromuscular assessment - NCS/EMG, acetylcholine receptor antibodies (myasthenia gravis)
- Polysomnography - if sleep-disordered breathing suspected
Treatment
1. Acute Respiratory Acidosis (Life-threatening)
- Reverse the underlying cause simultaneously with restoration of ventilation
- Tracheal intubation and mechanical ventilation if indicated
- Treat bronchospasm (acute asthma): nebulized bronchodilators, IV corticosteroids, magnesium sulfate
- Reverse opioid toxicity: naloxone (IV/IM/intranasal)
- Reverse benzodiazepine toxicity: flumazenil
2. Oxygen Therapy - Special Caution in COPD
- Must be titrated carefully in chronic CO₂ retainers
- Injudicious O₂ → removes hypoxic drive → further hypoventilation → worsening respiratory acidosis
- Target SpO₂: 88-92% in chronic hypercapnic COPD (not 94-98%)
- Non-invasive ventilation (NIV/BiPAP) is preferred over intubation in COPD exacerbation
3. Chronic Respiratory Acidosis
- Smoking cessation, bronchodilators, corticosteroids, diuretics, physiotherapy
- Long-term NIV (BiPAP) at home for obesity-hypoventilation syndrome, neuromuscular disease
- CPAP/BiPAP for obstructive sleep apnea
4. Correction of PaCO₂ - Important Caveat
- Never correct chronic hypercapnia rapidly
- Rapidly falling PaCO₂ → cardiac arrhythmias, seizures, reduced cerebral perfusion
- Lower PaCO₂ gradually to the patient's own baseline, not necessarily to 40 mmHg
- Provide adequate Cl⁻ and K⁺ to allow renal excretion of retained HCO₃⁻
5. Permissive Hypercapnia (in ARDS/ICU)
- Low tidal volume ventilation (6 mL/kg IBW) to prevent barotrauma
- Allows PaCO₂ to rise; acceptable pH target: approximately 7.20-7.25
- Sodium bicarbonate infusion may be needed if concurrent metabolic acidosis
- Absolute contraindication: raised intracranial pressure
Summary Table
| Feature | Detail |
|---|---|
| Primary disturbance | ↑ PaCO₂ (hypoventilation) |
| ABG pattern | pH ↓, PaCO₂ ↑, HCO₃⁻ ↑ |
| Compensation | Renal only (no respiratory compensation) |
| Acute HCO₃⁻ rise | 1 mEq/L per 10 mmHg ↑PaCO₂ |
| Chronic HCO₃⁻ rise | 4 mEq/L per 10 mmHg ↑PaCO₂ |
| Max HCO₃⁻ in compensation | ~38 mEq/L |
| Classic signs | Asterixis, papilledema, CO₂ narcosis |
| Most common chronic cause | COPD |
| Most common acute cause | Opioid overdose / acute severe asthma |
| O₂ caution | Target SpO₂ 88-92% in chronic CO₂ retainers |
| Correction caution | Never correct chronic hypercapnia rapidly |
Exam High-Yield Points
- Respiratory acidosis = hypoventilation = CO₂ retention
- No respiratory compensation - respiration IS the problem
- Acute vs. chronic distinction is the most important clinical differentiation
- Asterixis is the hallmark neurological sign of CO₂ retention
- Papilledema can occur due to CO₂-driven cerebral vasodilation
- Hypochloremia develops in chronic cases - kidneys excrete Cl⁻ to retain HCO₃⁻
- Hyperkalemia occurs due to H⁺/K⁺ exchange across cell membranes
- COPD + O₂: target 88-92%, not 94-98%
- Permissive hypercapnia in ARDS: deliberate strategy, pH > 7.20 acceptable
- If HCO₃⁻ too high → mixed with metabolic alkalosis (e.g., vomiting + COPD); if too low → mixed with metabolic acidosis (e.g., COPD + lactic acidosis from tissue hypoxia)
Sources: Brenner & Rector's The Kidney 10th Ed. | Costanzo Physiology 7th Ed. | Murray & Nadel's Textbook of Respiratory Medicine | Harrison's Principles of Internal Medicine 22nd Ed. | Miller's Anesthesia 10th Ed. | Sabiston Textbook of Surgery | Schwartz's Principles of Surgery
This is a shared conversation. Sign in to Orris to start your own chat.