Pathophysiology of acute pancreatitis
Reading File
Finding Sources
Searching PubMed
"acute pancreatitis"[MeSH Terms] AND pathophysiology
Finding Sources
Searching PubMed
"acute pancreatitis" AND "pathogenesis"
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Loading Image
Finding Sources
Reading File
This block is on pediatric pancreatitis. I have enough from Robbins and Rosen's for a complete pathophysiology answer. Let me now compile the comprehensive response.
Pathophysiology of Acute Pancreatitis
Acute pancreatitis is a reversible inflammatory disorder of the pancreas with severity ranging from focal edema and fat necrosis to widespread hemorrhagic necrosis. Its global incidence is 33-74 per 100,000 per year, with overall mortality around 5% (rising to ~30% in severe necrotizing forms). The fundamental mechanism is autodigestion - premature activation of proteolytic enzymes within the pancreatic parenchyma itself.
Etiology (Triggers)
Gallstones (~40%) and alcohol (~35%) account for close to 80% of cases. The remainder include:
| Category | Examples |
|---|---|
| Mechanical | Gallstones, pancreas divisum, biliary sludge, post-ERCP, trauma |
| Metabolic | Hypertriglyceridemia (>1000 mg/dL, 5-10% of cases), hypercalcemia |
| Toxic | Alcohol, drugs (azathioprine, valproate, thiazides, estrogens, chemotherapy) |
| Infectious | Mumps virus, coxsackievirus (direct acinar cell infection) |
| Genetic | PRSS1 (trypsinogen) mutations, SPINK1, CFTR mutations |
| Idiopathic | 10-20% of cases |
Core Pathogenic Mechanism: Premature Trypsin Activation
The central event is inappropriate intra-acinar activation of trypsinogen to trypsin. Under normal conditions, digestive proenzymes (zymogens) are synthesized in the endoplasmic reticulum, packaged in zymogen granules, and only activated in the duodenal lumen by enterokinase. Several mechanisms can disrupt this:
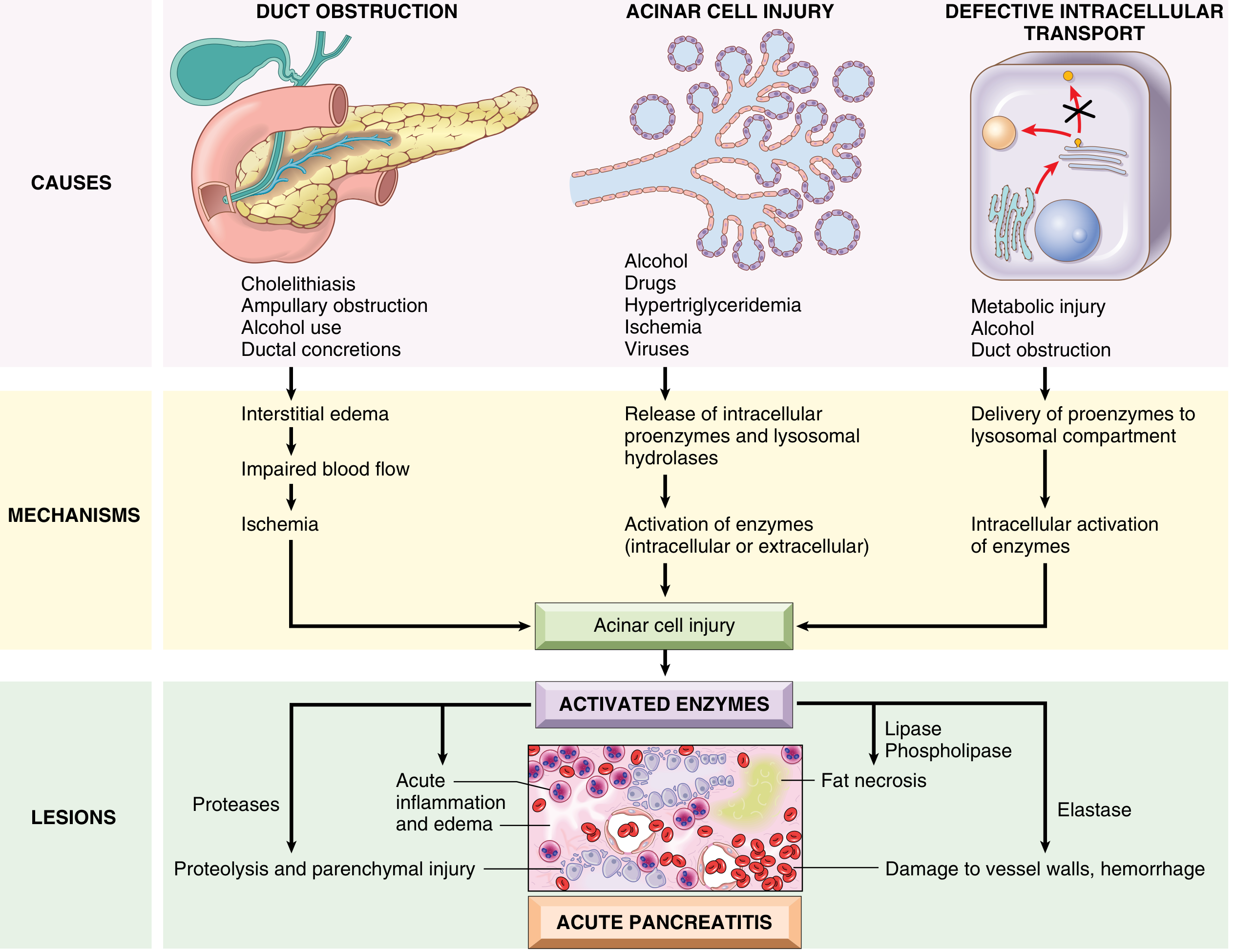
Fig. 15.2 - Proposed pathogenesis of acute pancreatitis (Robbins & Kumar Basic Pathology)
Three Initiating Pathways
1. Pancreatic Duct Obstruction
A gallstone or biliary sludge impacting at the ampulla of Vater blocks ductal outflow, raising intraductal pressure. Enzyme-rich interstitial fluid accumulates. Since lipase is secreted in active form, it immediately causes local fat necrosis. Injured tissue, periacinar myofibroblasts, and leukocytes release proinflammatory cytokines, promoting interstitial edema through a leaky microvasculature. The resulting edema further compresses local blood flow, causing ischemia and acinar cell injury.
2. Primary Acinar Cell Injury
Caused by alcohol, hypertriglyceridemia, ischemia, viral infections, and drugs. In hypertriglyceridemia, large triglyceride-rich chylomicrons impair capillary circulation, causing ischemic acinar injury. Injured cells release lipase into the interstitium, hydrolyzing triglycerides and liberating toxic free fatty acids. Alcohol acts via multiple mechanisms:
- Transiently increases exocrine secretion and causes sphincter of Oddi spasm
- Direct oxidative stress on acinar cells causing membrane damage
- Causes secretion of protein-rich pancreatic fluid, forming inspissated protein plugs that obstruct small ducts
- Triggers delivery of proenzymes to the lysosomal compartment (see below)
3. Defective Intracellular Transport of Proenzymes
In healthy acinar cells, digestive proenzymes and lysosomal hydrolases (e.g., cathepsin B) travel in completely separate intracellular pathways. In metabolic injury (alcohol, duct obstruction), these two compartments pathologically merge - proenzymes are co-delivered to the lysosomal compartment. Cathepsin B then cleaves trypsinogen to trypsin intracellularly, triggering a cascade of enzyme activation before any material reaches the duodenum.
Trypsin as the Master Activator
Once trypsin is abnormally active, it:
- Activates all other digestive proenzymes (chymotrypsinogen, proelastase, prophospholipase A2, prolipase) - triggering a positive feedback loop
- Converts prekallikrein → kallikrein, initiating the kinin system (bradykinin release → vasodilation and pain)
- Activates Factor XII (Hageman factor), triggering the clotting cascade and complement system
- Directly injures blood vessel walls, enabling hemorrhage
In hereditary pancreatitis, PRSS1 mutations eliminate the self-cleavage inactivation site of trypsin, abolishing a key negative feedback mechanism and leading to persistent hyperactive trypsin.
Downstream Cascade of Injury
Once enzymes are activated, a self-amplifying destructive sequence unfolds:
Trypsin activation
↓
Proteases → proteolytic destruction of parenchyma
Lipase/Phospholipase A2 → fat necrosis (saponification with Ca²⁺)
Elastase → dissolution of vessel walls → hemorrhage
↓
Macrophage/neutrophil recruitment
↓
Cytokine storm (IL-1β, IL-6, IL-8, TNF-α)
↓
Increased vascular permeability → edema, third-spacing
↓
Systemic Inflammatory Response Syndrome (SIRS)
Local Pathological Consequences
The four basic morphological alterations (Robbins) are:
- Microvascular leakage - causing interstitial edema
- Fat necrosis by lipases - released fatty acids + calcium → chalky white calcium soaps
- Acute inflammatory reaction - neutrophil/macrophage infiltration
- Proteolytic destruction of parenchyma and blood vessels → hemorrhage
Interstitial edematous pancreatitis (~90-95% of cases): focal edema and scattered fat necrosis; usually resolves within the first week.
Necrotizing pancreatitis (~5-10%): damage extends to acinar cells, ductal cells, islets of Langerhans, and blood vessels. Macroscopically: red-black hemorrhagic areas interspersed with yellow-white chalky fat necrosis foci. Fat necrosis can extend to omentum, bowel mesentery, and subcutaneous fat.
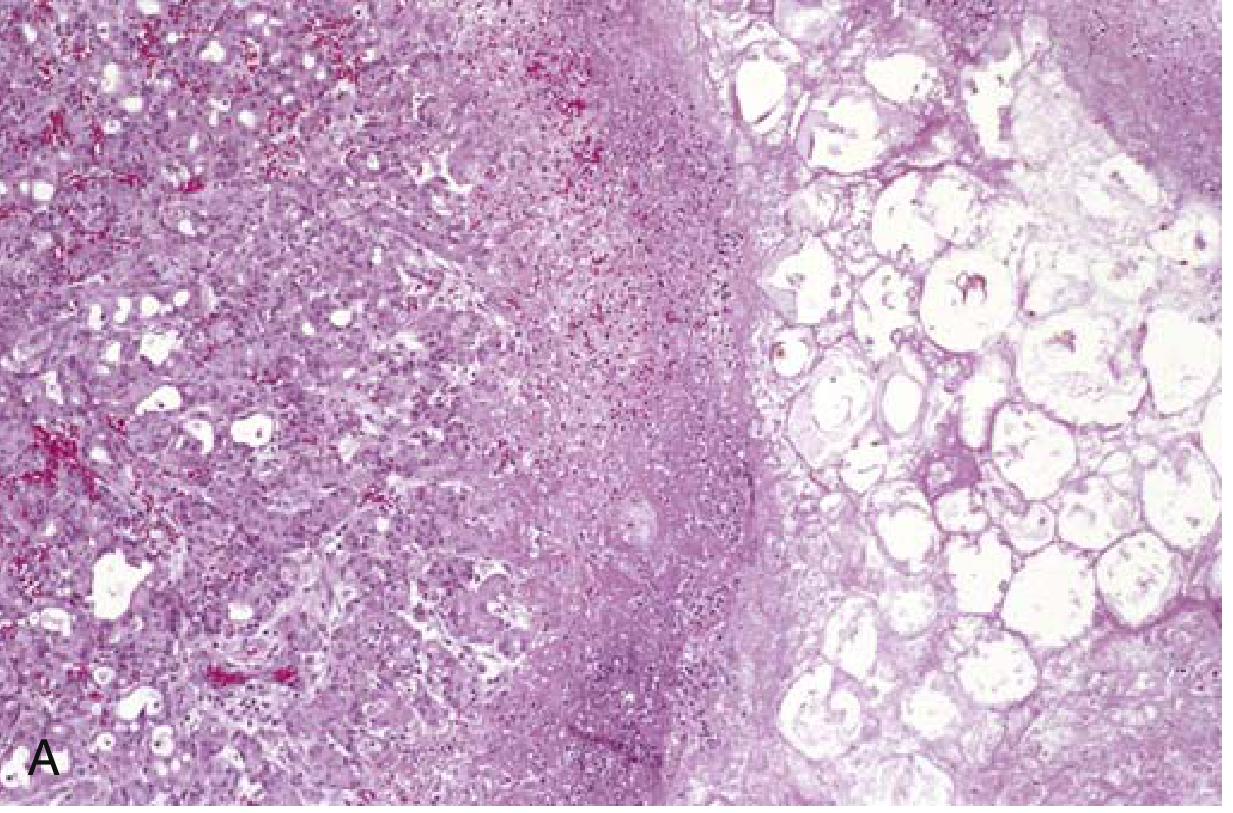
Fig. 15.3A - Acute necrotizing pancreatitis: necrotic parenchyma (left) abutting fat cells undergoing necrosis (right)
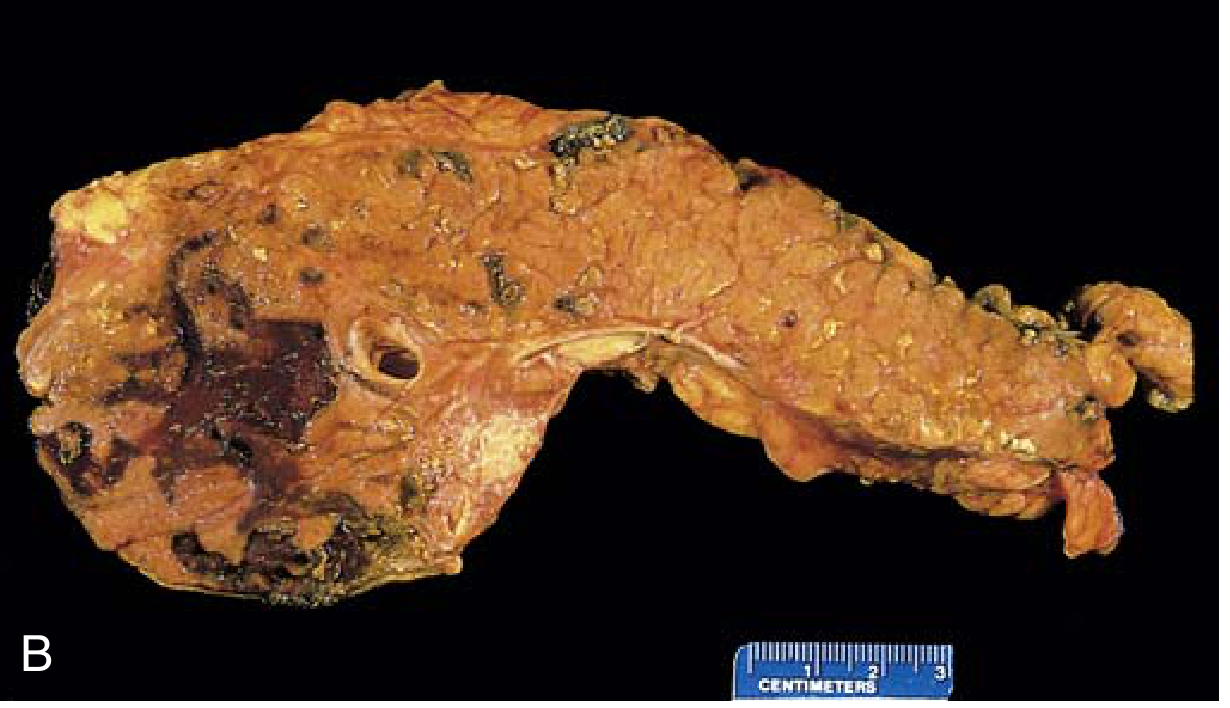
Fig. 15.3B - Gross specimen of hemorrhagic pancreatitis: dark hemorrhagic areas and chalky fat necrosis deposits
Systemic Complications (SIRS to MOF)
If the local inflammatory response overwhelms containment mechanisms, it spills systemically:
- Pulmonary: increased microvascular permeability + enzymatic degradation of surfactant (phospholipase A2) → ARDS, pleural effusions (left > right, present in ~50% of severe cases), atelectasis
- Cardiovascular: massive fluid third-spacing + vasodilation → distributive shock, decreased MAP
- Renal: hypoperfusion + inflammatory mediators → acute tubular necrosis / renal failure
- Coagulopathy: cytokine-mediated coagulation cascade activation → DIC
- Sepsis: bacterial translocation across the disrupted gut mucosal barrier → bacteremia and infected pancreatic necrosis
- Metabolic: hypocalcemia (from saponification), hyperglycemia (islet destruction)
Grey Turner sign (flank discoloration) and Cullen sign (periumbilical discoloration) reflect retroperitoneal and intraperitoneal hemorrhage respectively - rare but prognostically ominous signs.
Summary Flowchart
Inciting event (gallstone / alcohol / hypertriglyceridemia / drugs)
↓
Acinar cell injury (via duct obstruction / direct toxicity / defective transport)
↓
Premature trypsinogen → trypsin activation
↓
Cascade activation of all digestive enzymes
↓
Autodigestion: fat necrosis, proteolysis, hemorrhage
↓
Cytokine release (TNF-α, IL-1β, IL-6), complement, kinin activation
↓
Local: edema, interstitial pancreatitis → necrotizing pancreatitis
↓
Systemic: SIRS → ARDS, renal failure, shock, DIC (multi-organ failure)
Sources:
- Robbins & Kumar Basic Pathology, Chapter 15 (Pancreas), pp. 640-641
- Rosen's Emergency Medicine, Chapter 77 (Pancreatitis), pp. 1264-1265
- Sleisenger & Fordtran's Gastrointestinal and Liver Disease, Chapter 57
Management of shock
Reading File
Finding Sources
Searching PubMed
"shock" AND "management" AND "resuscitation"
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive, high-quality information from multiple authoritative textbooks. I have enough to write a thorough, well-structured answer.
Management of Shock
Shock is a clinical syndrome of inadequate tissue perfusion - a life-threatening failure of the circulatory system to deliver sufficient oxygen and nutrients to cells. At the cellular level, shock first impairs mitochondrial function and aerobic metabolism, leading to anaerobic glycolysis, lactic acid accumulation, and eventually cell death if uncorrected.
Classification of Shock
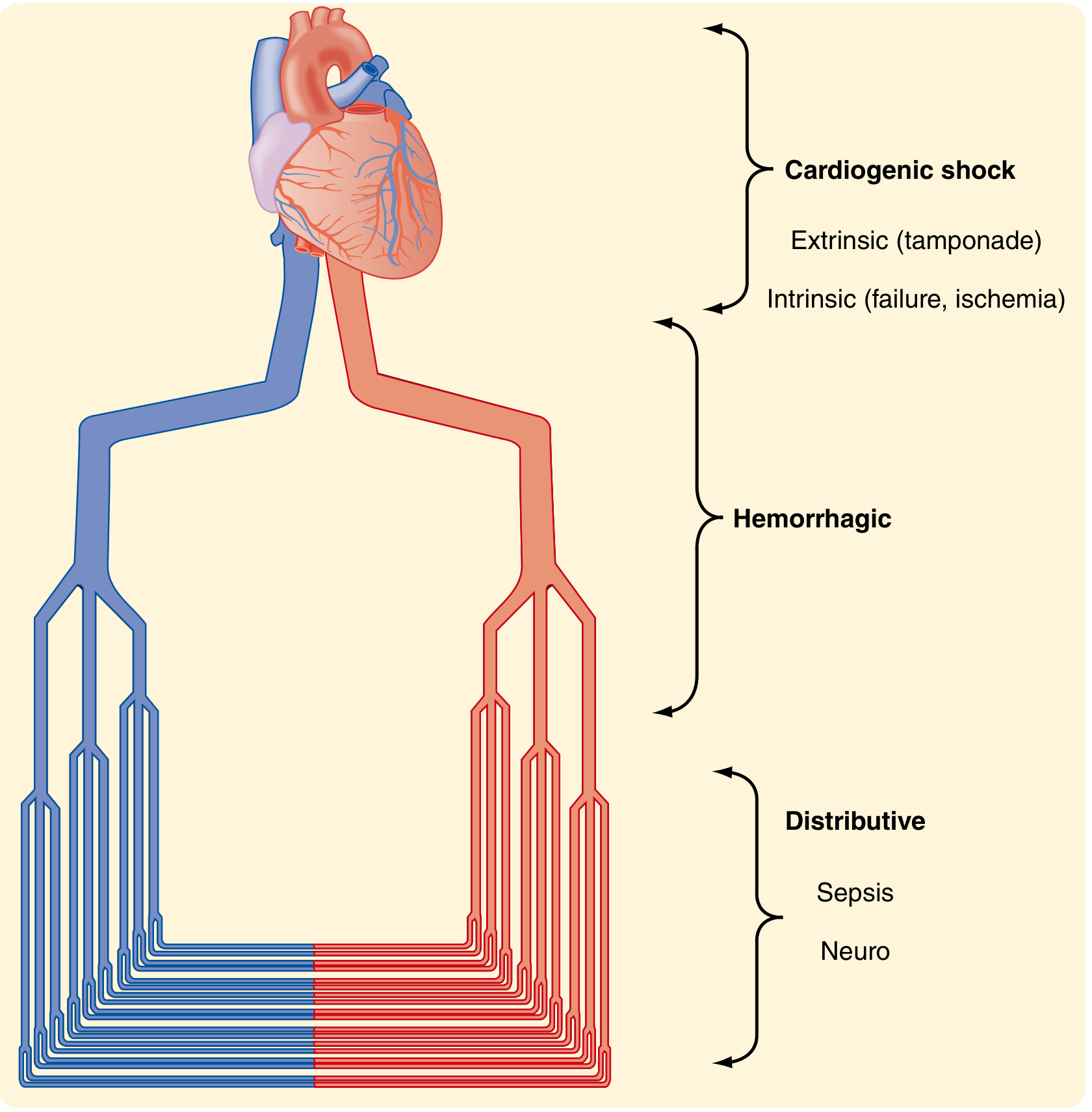
Fig. 33.5 - Types of Shock (Sabiston Textbook of Surgery)
| Type | Mechanism | Common Causes |
|---|---|---|
| Hypovolemic/Hemorrhagic | Reduced preload from volume loss | Hemorrhage, burns, dehydration, GI losses |
| Cardiogenic | Pump failure (intrinsic or extrinsic) | MI, acute heart failure, cardiac tamponade, tension pneumothorax, PE |
| Distributive | Pathologic vasodilation, maldistribution of flow | Sepsis, anaphylaxis, neurogenic (spinal injury) |
| Obstructive | Mechanical obstruction to cardiac output | Massive PE, cardiac tamponade, tension pneumothorax |
Key concept: Shock can exist with a normal blood pressure. A base deficit more negative than -4 mEq/L or serum lactate >4.0 mmol/L warrants a presumptive diagnosis of shock regardless of BP.
General Principles of Management (All Shock Types)
Step 1: Secure Airway, Breathing, Circulation
- High-flow oxygen; early intubation if respiratory failure or severe obtundation
- Two large-bore IV lines (minimum 16G); intraosseous (IO) access if IV access is delayed - IO can deliver all resuscitation fluids and medications
- If IV/IO unavailable, peripherally-sited vasopressors can be infused via an 18G or larger catheter at the antecubital fossa or more proximally
Step 2: Identify and Treat the Underlying Cause Simultaneously
This is non-negotiable. Resuscitation without source control is insufficient:
- Hemorrhagic: control bleeding
- Septic: antibiotics + source control
- Cardiogenic: revascularization / mechanical support
- Obstructive: decompress (needle thoracostomy, pericardiocentesis, thrombolysis)
Step 3: Hemodynamic Monitoring and Targets
- Urine output: target >0.5 mL/kg/h (reliable index of renal perfusion; normal is 1.0 mL/kg/h)
- Serum lactate: trend is more important than single value; lactate clearance >10% per 2h is a positive sign
- Base deficit: target toward 0; base deficit worse than -4 mEq/L = significant shock
- MAP: generally target ≥65 mmHg
- Arterial line for continuous BP monitoring in refractory shock
- Central venous access for vasopressor infusion and CVP monitoring when peripheral access is inadequate
1. Hemorrhagic Shock (Hypovolemic)
ATLS Classification
| Class | Blood Loss | HR | BP | RR | Urine (mL/h) | CNS |
|---|---|---|---|---|---|---|
| I | 0-15% | <100 | Normal | 14-20 | >30 | Slightly anxious |
| II | 15-30% | >100 | Normal | 20-30 | 20-30 | Mildly anxious |
| III | 30-40% | >120 | Decreased | 30-40 | 5-15 | Anxious/confused |
| IV | >40% | >140 | Decreased | >35 | Negligible | Confused/lethargic |
Caveat: ATLS classes are not rigidly reliable. Tachycardia is neither sensitive nor specific for hemorrhage. Children compensate with large volumes until sudden decompensation; elderly patients decompensate at lower volumes.
Resuscitation Strategy
-
Immediate hemorrhage control: direct pressure, tourniquet, traction splinting for long-bone fractures, REBOA (resuscitative endovascular balloon occlusion of the aorta) for non-compressible torso hemorrhage
-
Fluid resuscitation:
- Class I-II: judicious isotonic crystalloid (10-20 mL/kg)
- Class III-IV or suspected massive hemorrhage: balanced transfusion of packed red blood cells (PRBCs) : fresh frozen plasma (FFP) : platelets in a 1:1:1 ratio is preferred over crystalloid alone
- Avoid large-volume crystalloid resuscitation - causes dilutional coagulopathy, hypothermia, and abdominal compartment syndrome
-
Permissive hypotension (damage control resuscitation): in penetrating trauma with uncontrolled bleeding, target SBP ~80-90 mmHg until hemorrhage control is achieved - prevents "popping the clot"
-
Lethal Triad prevention: Hypothermia + Acidosis + Coagulopathy form a deadly positive-feedback loop:
- Hypothermia impairs coagulation enzyme activity by 40% even with a drop of a few degrees
- Warm all IV fluids; use warming blankets; minimize heat loss
- Transfuse early to correct coagulopathy
-
Transfusion trigger: Hgb <7 g/dL in stable patients; higher thresholds in active hemorrhage or cardiac ischemia
2. Septic Shock (Distributive)
Definition (Sepsis-3)
- Sepsis: suspected/confirmed infection + SOFA score increase ≥2
- Septic shock: sepsis + vasopressor requirement + lactate >2 mmol/L despite adequate volume
Hour-by-Hour Management (Surviving Sepsis Bundle)
Within 1 hour:
- Obtain blood cultures (≥2 sets) before starting antibiotics
- Administer broad-spectrum IV antibiotics within 1 hour of sepsis recognition
- Measure lactate
- Begin IV fluid resuscitation
Fluid Resuscitation:
- Initial bolus: 30 mL/kg IV balanced crystalloid (e.g., lactated Ringer's preferred over 0.9% NaCl, which causes hyperchloremic acidosis) in the first 3 hours
- Reassess frequently with dynamic indices (pulse pressure variation, stroke volume variation) - avoid fluid overload
- Add albumin when large volumes of crystalloid are required
- Do not use hetastarch (HES) - associated with acute kidney injury and increased mortality
Vasopressors (when MAP <65 mmHg despite adequate fluids):
- First-line: Norepinephrine (0.01-3 mcg/kg/min) - α1 and β1 agonist; increases SVR and MAP
- Vasopressin: add as fixed-rate adjunct (0.03-0.04 units/min) when norepinephrine dose reaches 0.25-0.5 mcg/kg/min; do not use alone
- Epinephrine: add if hypotension persists despite NE + vasopressin
- Dopamine: avoid in most cases; reserve for highly selected circumstances (e.g., bradycardic patients)
- Dobutamine: add to norepinephrine if low cardiac output state persists despite adequate fluid resuscitation
Corticosteroids:
- Hydrocortisone 200 mg/day IV (as continuous infusion or divided doses) if shock persists despite adequate fluids and vasopressors
- Do not use to treat sepsis without shock
Supportive Care:
- Target Hgb 7-9 g/dL (higher only in tissue hypoperfusion, CAD, or acute hemorrhage)
- For ARDS: low tidal volume ventilation (6 mL/kg ideal body weight), plateau pressure ≤30 cmH2O, PEEP titration
- Prone positioning ≥12 hours/day for moderate-severe ARDS
- ECMO if severe ARDS fails conventional ventilation (in experienced centers)
- Tight glucose control: avoid hypoglycemia; target <180 mg/dL
3. Cardiogenic Shock
Key Features
- Low cardiac output + elevated filling pressures
- Clinical: cold, clammy extremities; elevated JVP; pulmonary oedema; S3 gallop
Management
- Oxygen and ventilatory support: non-invasive positive pressure ventilation (CPAP/BiPAP) reduces work of breathing and preload in pulmonary oedema; intubate if needed; apply PEEP
- Treat the underlying cause urgently:
- ACS: emergent PCI (percutaneous coronary intervention) - time-critical
- Cardiac tamponade: pericardiocentesis
- Tension pneumothorax: needle decompression
- Massive PE: systemic thrombolysis or catheter-directed therapy
- Vasopressor/Inotrope selection:
- Norepinephrine (0.5 mcg/min starting): first-line vasopressor for MAP support
- Dobutamine (5 mcg/kg/min starting): positive inotrope; reduces afterload - add to NE in low-output states
- Caution: dopamine increases arrhythmia risk; epinephrine increases myocardial O2 demand; vasodilators (nitroprusside) may be used if adequate MAP maintained to reduce afterload
- Mechanical circulatory support (MCS) for refractory shock:
- Intra-aortic balloon pump (IABP): increases coronary perfusion, reduces afterload
- Impella device / ventricular assist devices (VAD): more powerful LV unloading
- Veno-arterial ECMO (VA-ECMO): for refractory cardiogenic shock in experienced centers
- Avoid excessive IV fluids: cardiogenic shock is preload-sensitive but fluid-overload worsens pulmonary oedema
4. Obstructive Shock
Requires immediate mechanical relief of the obstruction - vasopressors and fluids are temporizing only:
| Cause | Immediate Treatment |
|---|---|
| Tension pneumothorax | Needle decompression (2nd ICS MCL) → chest tube |
| Cardiac tamponade | Pericardiocentesis; pericardial window |
| Massive PE | Anticoagulation; systemic thrombolysis (tPA); catheter-directed therapy; surgical embolectomy |
| Aortic dissection | Surgical emergency; beta-blockers to reduce aortic wall stress |
5. Neurogenic Shock (Distributive Subtype)
- Caused by loss of sympathetic tone below spinal cord injury (T6 or above)
- Presents with hypotension + bradycardia (distinguishes from other shock types) + warm, dry skin
- Management:
- IV fluid resuscitation
- Vasopressors (norepinephrine or phenylephrine) to restore vascular tone
- Atropine or cardiac pacing if bradycardia is symptomatic
- Maintain spine immobilization
6. Anaphylactic Shock
- Epinephrine IM (0.3-0.5 mg of 1:1000) into the lateral thigh is the cornerstone - given immediately, not after other drugs
- IV access; 1-2 L crystalloid for hypotension
- H1 + H2 antihistamines (adjuncts only - do not delay epinephrine)
- Corticosteroids (prevent biphasic reaction)
- Nebulized bronchodilators for bronchospasm
- Vasopressors if hypotension refractory to epinephrine
End-Points of Resuscitation
A combination of the following markers guides adequacy of resuscitation:
| Parameter | Target |
|---|---|
| MAP | ≥65 mmHg |
| Urine output | >0.5 mL/kg/h |
| Serum lactate | Clearance >10%/2h; normalization <2 mmol/L |
| Base deficit | Trend toward 0 (> -4 mEq/L) |
| Central venous O2 sat (ScvO2) | >70% (adjunct; not mandated) |
Note: Early Goal-Directed Therapy (EGDT) using strict CVP and ScvO2 targets has not demonstrated mortality benefit over modern protocolized care in large trials (ProCESS, ARISE, ProMISe). Current emphasis is on early recognition, early antibiotics, early fluids, and close monitoring rather than invasive haemodynamic targets.
Sources:
- Rosen's Emergency Medicine, Chapter 3 (Shock), pp. 59-63
- Sabiston Textbook of Surgery (19e), Chapter 33 (Shock), pp. 563-572
- Goodman & Gilman's Pharmacological Basis of Therapeutics, Chapter - Shock, pp. 285-286
- Barash Clinical Anesthesia (9e), Table 57-4 (Management of Sepsis and Septic Shock), pp. 4831+
This is a shared conversation. Sign in to Orris to start your own chat.