hepatopulmonary syndrome pathophysiology
Reading File
Finding Sources
Searching PubMed
"hepatopulmonary syndrome" AND pathophysiology
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive information from two authoritative textbooks and current PubMed reviews. Here is the full answer:
Hepatopulmonary Syndrome (HPS) - Pathophysiology
Definition
HPS is defined by a widened alveolar-arterial oxygen gradient (AaPO2) on room air - normal is <15 mmHg (<20 mmHg in those ≥64 years) - resulting from intrapulmonary vascular dilatation in the setting of advanced liver disease or portal hypertension, with or without overt hypoxemia. Severity is graded by PaO2: mild (≥80), moderate (>60-80), severe (50-60), very severe (<50 mmHg). - Sleisenger and Fordtran's GI and Liver Disease, p. 1821
Core Pathophysiologic Cascade
The central lesion is pulmonary microvascular dilatation (precapillary and capillary) driven by loss of normal vasoregulatory control. The cascade, illustrated below, proceeds as follows:
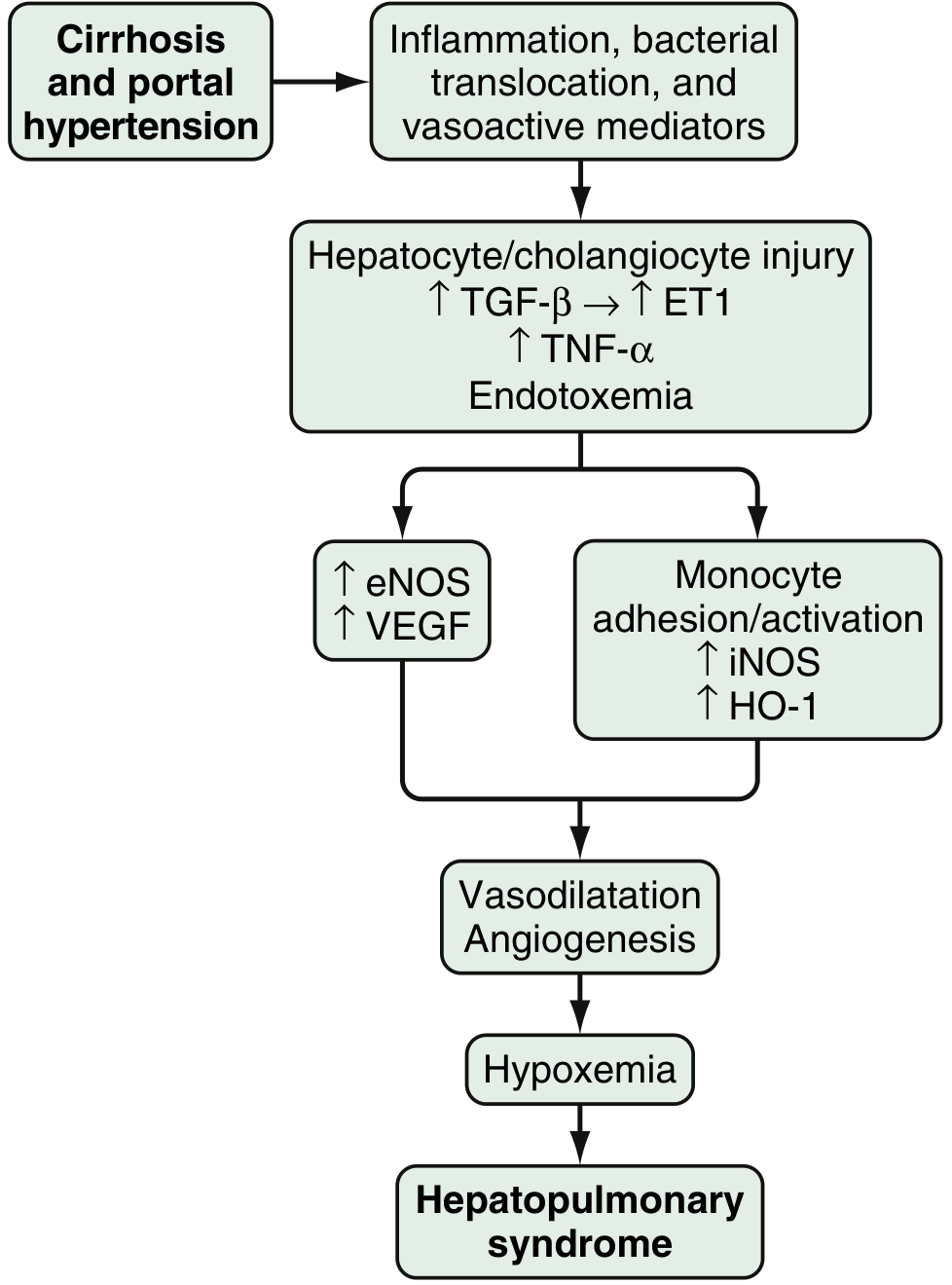
Fig. 94.3 - Sleisenger and Fordtran's GI and Liver Disease
Step 1: Initiating Signal from the Diseased Liver
Cirrhosis/portal hypertension drives:
- Bacterial translocation from the gut into portal circulation
- Release of endothelin-1 (ET-1) via hepatocyte/cholangiocyte injury (mediated by TGF-β → ET-1)
- Elevated circulating TNF-α and endotoxemia
Step 2: Two Parallel Vasoactive Arms in the Pulmonary Microvasculature
Arm 1 - eNOS/Endothelin-B axis:
- Shear stress in the pulmonary vasculature upregulates endothelin-B receptors on pulmonary endothelium
- ET-1 acting on ET-B receptors activates endothelial NOS (eNOS), producing excess nitric oxide (NO)
- VEGF is also upregulated, contributing to angiogenesis
Arm 2 - Monocyte/macrophage adhesion:
- ET-B receptor activation, fractalkine receptor overexpression, and TNF-α drive monocyte/macrophage adhesion within the pulmonary microvasculature
- Adherent macrophages activate:
- iNOS → additional NO production
- Heme oxygenase-1 (HO-1) → carbon monoxide (CO) production (another potent vasodilator)
- VEGF signaling → vascular angiogenesis
Both arms converge on pulmonary vasodilatation and angiogenesis.
Three Mechanisms of Hypoxemia
The resulting vascular changes impair oxygenation through three overlapping mechanisms: - Murray & Nadel's Textbook of Respiratory Medicine, p. 962
| Mechanism | Explanation | Evidence |
|---|---|---|
| Low V/Q ratio | Most common mechanism; dilated vessels receive blood that bypasses well-ventilated alveoli; explains frequent response to supplemental O2; worsens in upright position (orthodeoxia) due to increased basilar perfusion | MIGET studies confirm increased perfusion of low-V/Q units |
| Diffusion limitation | Capillary dilatation + high cardiac output (typical in cirrhosis) reduce RBC transit time; dilated vessels place erythrocytes in the center of the lumen, far from the alveolar gas, widening diffusion distance | Consistent with O2 responsiveness; not definitively confirmed by MIGET |
| Intrapulmonary shunt | Dominant in severe disease (PaO2 35-67 mmHg); macroaggregated albumin and bubble echocardiography demonstrate large-particle transit through lung; paradoxically, even shunt areas may partially respond to 100% O2, behaving like extreme low-V/Q | Tc-99m-MAA shunt fraction (~32%) often exceeds 100% O2 shunt estimate (~19%), indicating intermediate behavior |
Key point on shunt behavior: In HPS, vascular shunts detected by contrast echo or radionuclide methods can simultaneously behave as pure shunts (non-responsive to O2) or as areas of diffusion limitation/low-V/Q (partially responsive to O2), depending on vessel size and location. This explains the apparent contradiction of "shunt" that improves with supplemental oxygen.
Why Orthodeoxia Occurs
The dilatations are preferentially in the lower lung zones (basilar predominance). In the upright position, gravity directs more blood to these dilated, mismatch-prone zones, worsening hypoxemia. Lying down redistributes flow, improving oxygenation - producing the pathognomonic platypnea-orthodeoxia phenomenon.
Role of Angiogenesis
Beyond simple vasodilation, new vessel formation contributes to the chronicity and severity of HPS. VEGF (via VEGF receptor signaling and placental growth factor in mouse models) drives structural remodeling. This explains why:
- HPS may take >1 year to fully resolve after liver transplantation even though vasoregulation can normalize quickly
- Single nucleotide polymorphisms in angiogenic genes are more prevalent in HPS patients than in cirrhotic patients without HPS - Sleisenger and Fordtran, p. 1821
Summary Pathway
Cirrhosis/Portal HTN
↓
Bacterial translocation + hepatocyte injury
↓
↑ ET-1, TNF-α, endotoxemia
↓
┌────────────────────────────────────┐
│ Pulmonary endothelium │
│ ET-B receptor overexpression │
│ Fractalkine receptor upregulation │
└────────────────────────────────────┘
↓ ↓
eNOS activation Monocyte adhesion
VEGF signaling → iNOS activation
→ HO-1 activation
↓ ↓
NO excess NO + CO excess
↓
Precapillary/capillary dilatation
+ Angiogenesis
↓
Low V/Q + Diffusion limitation + Shunt
↓
HYPOXEMIA (HPS)
Recent Literature
Recent reviews (2024-2025) continue to refine understanding of HPS:
- Bommena & Fallon (2024) reviewed pulmonary complications of portal hypertension, contextualizing HPS within the spectrum of portopulmonary vascular disease (PMID: 38945638)
- La Via et al. (2025) specifically reviewed HPS pathophysiological mechanisms and clinical implications for anesthesiology (PMID: 40493793)
- Medina-Morales & Schiano (2025) provides an updated comparison of HPS vs portopulmonary hypertension (PMID: 40670035)
No recent meta-analyses contradicting the textbook framework were identified.
This is a shared conversation. Sign in to Orris to start your own chat.