Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive mechanistic content from Harrison's, Goldman-Cecil, Robbins, Murray & Nadel, and Sabiston. I have two excellent diagrams. I have enough to write a thorough, well-structured answer.
Acute Respiratory Distress Syndrome (ARDS) - Mechanism
Definition and Berlin Criteria
ARDS is a clinical syndrome of acute hypoxemic respiratory failure caused by diffuse alveolar damage (DAD). The Berlin definition requires:
- Bilateral opacities on chest X-ray or CT not explained by effusions, lobar collapse, or nodules
- PaO₂/FiO₂ ratio <300 mmHg on PEEP ≥5 cm H₂O (mild <300, moderate <200, severe <100)
- Onset within 7 days of a known clinical insult
- Respiratory failure not fully explained by heart failure or fluid overload
Common triggers include sepsis, severe trauma, aspiration, diffuse pneumonia, pancreatitis, and blood product transfusions (Sabiston Textbook of Surgery, p. 940).
Time Course of ARDS

ARDS progresses from alveolar edema (days 0-2) → hyaline membrane formation (by day 7) → interstitial inflammation (days 7-21) → fibrosis in some patients (>21 days) (Harrison's Principles of Internal Medicine 22E, Fig. 312-1)
Phase 1 - Exudative Phase (Days 0-7)
This is the acute injury phase. The pathological cascade proceeds as follows:
1. Initial Insult and Barrier Disruption
An inciting event (direct lung injury or systemic insult) damages alveolar capillary endothelial cells and type I pneumocytes (which form ~95% of the alveolar surface). The normally tight alveolar barrier fails, allowing protein-rich fluid to flood the interstitial and alveolar spaces - producing non-cardiogenic pulmonary edema (Harrison's, p. 2343).
2. Toll-Like Receptor Activation and Cytokine Storm
Bacteria, viruses, or DAMPs (damage-associated molecular patterns) activate Toll-like receptors on resident alveolar macrophages and type I pneumocytes. This triggers secretion of proinflammatory cytokines:
- IL-1, IL-6, IL-8, TNF-α - drive systemic and local inflammation
- Leukotriene B₂ - potent neutrophil chemoattractant
These mediators massively recruit circulating leukocytes, especially neutrophils, into the pulmonary interstitium and alveoli (Harrison's, p. 2343).
3. Neutrophil-Mediated Injury (Central Mechanism)
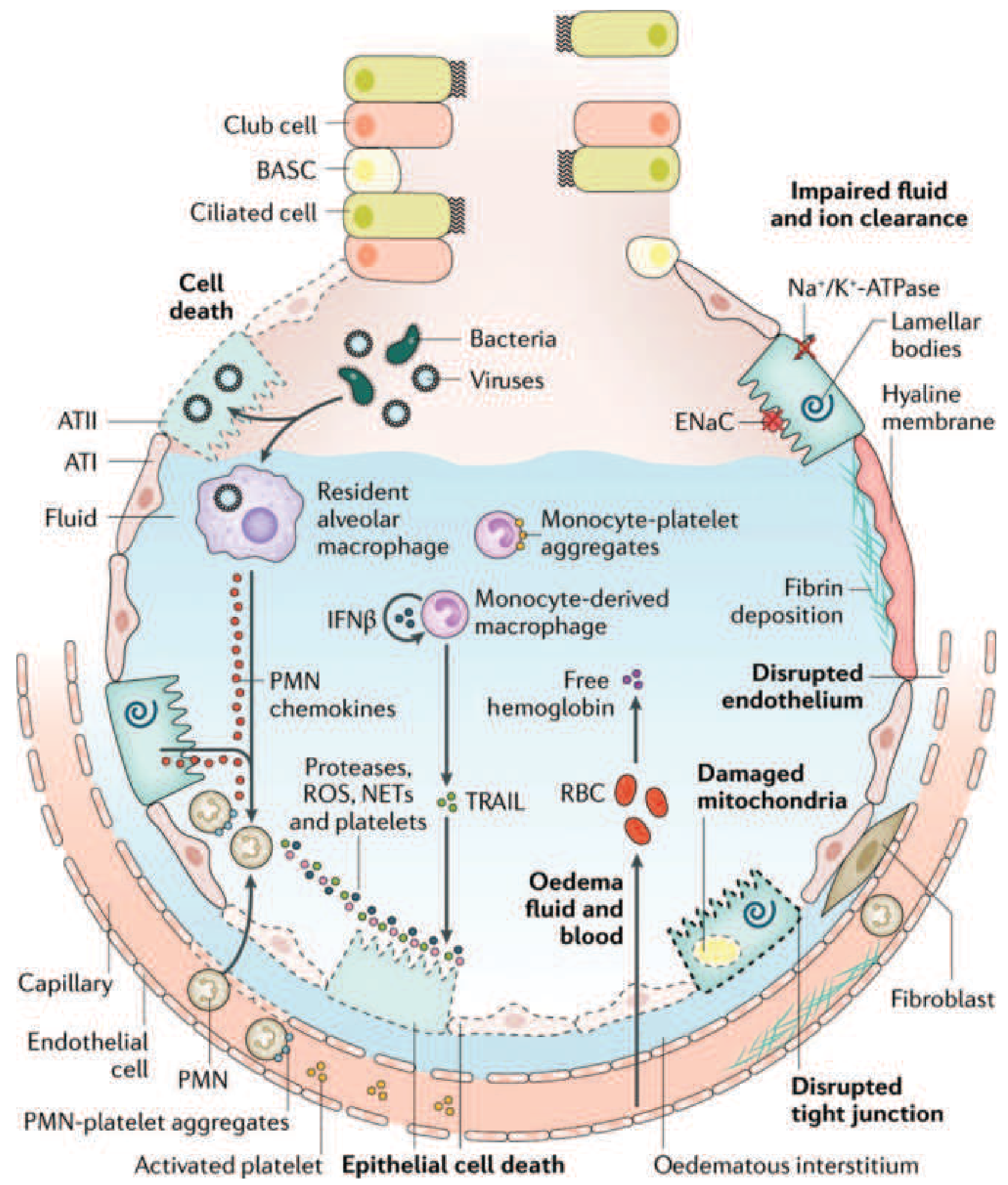
The injured alveolus in acute ARDS. Neutrophils (PMNs) migrate across the epithelium and release proteases, reactive oxygen species (ROS), and neutrophil extracellular traps (NETs). Monocyte-derived macrophages contribute via TRAIL-induced epithelial apoptosis. Na⁺/K⁺-ATPase and ENaC on type II cells are disabled, impairing fluid clearance. Hyaline membranes form on the denuded epithelial surface (Harrison's, Fig. 312-3)
Activated neutrophils that have transmigrated into the alveolar space release:
- Proteases (elastase, MMP-9) - digest extracellular matrix and endothelial/epithelial junctions
- Reactive oxygen species (ROS) - oxidative injury to cell membranes and proteins
- Neutrophil extracellular traps (NETs) - chromatin-platelet aggregates that amplify injury
- Phospholipase A₂ - degrades surfactant phospholipids, causing alveolar collapse and increased vascular permeability (Murray & Nadel's, block29, p. 2961)
4. Surfactant Dysfunction
Type II pneumocytes are damaged, reducing surfactant synthesis. What surfactant remains is degraded by phospholipase A₂ and plasma proteins flooding the airspace. Loss of surfactant raises alveolar surface tension, causing:
- Diffuse alveolar collapse (atelectasis)
- Reduced lung compliance
- Worsened intrapulmonary shunting
5. Hyaline Membrane Formation
Condensed plasma proteins (fibrin, albumin) aggregate in the air spaces along with cellular debris and dysfunctional surfactant, forming hyaline membrane whorls along denuded alveolar walls - the pathological hallmark of DAD (Harrison's, p. 2343).
6. Pulmonary Vascular Injury
Vascular injury occurs simultaneously:
- Microthrombi form in pulmonary capillaries (fibrocellular proliferation, monocyte-platelet aggregates, activated platelets)
- Vascular obliteration raises pulmonary vascular resistance and causes pulmonary hypertension
- Blood flow is redirected away from ventilated alveoli, increasing dead space
- The combination of shunting (blood past unventilated alveoli) and dead space (ventilation without perfusion) produces severe hypoxemia and hypercapnia (Harrison's, p. 2344)
7. Gravity-Dependent Distribution
The heavy, edematous lung behaves like a waterlogged sponge. Edema and atelectasis predominate in dependent zones (posterior in the supine patient). CT demonstrates striking heterogeneity: consolidated dependent regions, ground-glass intermediate zones, and relatively spared non-dependent areas. This heterogeneity is why high tidal volumes injure the "baby lung" of remaining normal tissue (Goldman-Cecil, p. 1067).
Phase 2 - Proliferative Phase (Days 7-21)
Many patients begin recovering during this phase, but pathological changes continue:
- Lung repair is initiated: type II pneumocytes proliferate along denuded alveolar basement membranes
- They synthesize new surfactant and differentiate into type I pneumocytes
- A lymphocyte-predominant infiltrate replaces the earlier neutrophilic one
- Alveolar exudates begin to organize
- Interstitial inflammation and early fibrosis develop in some patients
Patients may still have dyspnea, tachypnea, and hypoxemia at this stage despite histological signs of resolution (Harrison's, p. 2344).
Phase 3 - Fibrotic Phase (>21 Days)
A subset of patients fail to resolve and enter this phase:
- Alveolar edema and inflammatory exudates convert to dense alveolar-duct and interstitial fibrosis
- Disrupted acinar architecture leads to emphysema-like bullae
- Intimal fibroproliferation in the pulmonary microcirculation causes progressive vascular occlusion and pulmonary hypertension
- Consequences include reduced compliance, increased dead space, and increased pneumothorax risk
- Biopsy evidence of fibrosis at any stage is associated with increased mortality (Harrison's, p. 2345)
Summary of Key Mediators
| Mediator | Source | Effect |
|---|---|---|
| TNF-α, IL-1, IL-6 | Macrophages | Systemic inflammation, neutrophil recruitment |
| IL-8 | Macrophages, epithelium | Neutrophil chemotaxis |
| Leukotriene B₂ | Multiple | Neutrophil recruitment |
| Elastase, MMP-9 | Neutrophils | Barrier destruction |
| ROS | Neutrophils | Oxidative cell injury |
| NETs | Neutrophils | Amplify inflammation and coagulation |
| Phospholipase A₂ | Pancreas/neutrophils | Surfactant degradation |
| TRAIL | Monocyte-derived macrophages | Epithelial apoptosis |
| Activated platelets | Circulation | Microvascular thrombosis, amplify inflammation |
Physiological Consequences
- Intrapulmonary shunting - blood passes through non-ventilated, consolidated alveoli → hypoxemia refractory to supplemental oxygen
- Increased dead space - ventilated alveoli with no perfusion (microthrombi) → hypercapnia
- Reduced compliance - stiff, edematous lung requires high airway pressures → barotrauma/volutrauma risk
- Pulmonary hypertension - vascular obliteration + hypoxic vasoconstriction → right heart strain
Sources
- Harrison's Principles of Internal Medicine 22E, Chapter 312, pp. 2343-2345
- Murray & Nadel's Textbook of Respiratory Medicine, Block 29 (pancreatitis-associated ARDS)
- Goldman-Cecil Medicine, Chapters 90-91, pp. 1066-1067
- Sabiston Textbook of Surgery, Chapter on Critical Care, p. 940
- Robbins & Kumar Basic Pathology, Acute Lung Injury and ARDS section, p. 1741
This is a shared conversation. Sign in to Orris to start your own chat.