Azotemia
Reading File
Finding Sources
Finding Sources
Searching PubMed
azotemia acute kidney injury
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have all the essential content to write a comprehensive answer on azotemia. Let me compile this into a thorough response.
Azotemia
Azotemia (from the Greek "azo" = nitrogen + "-emia" = in the blood) refers to the retention of nitrogenous waste products - primarily urea and creatinine - in the blood due to a reduced glomerular filtration rate (GFR). It is a laboratory finding, not a clinical diagnosis, and is the hallmark of acute kidney injury (AKI) and chronic kidney disease (CKD).
Classification
Azotemia is classified into three categories based on the site of the primary problem:
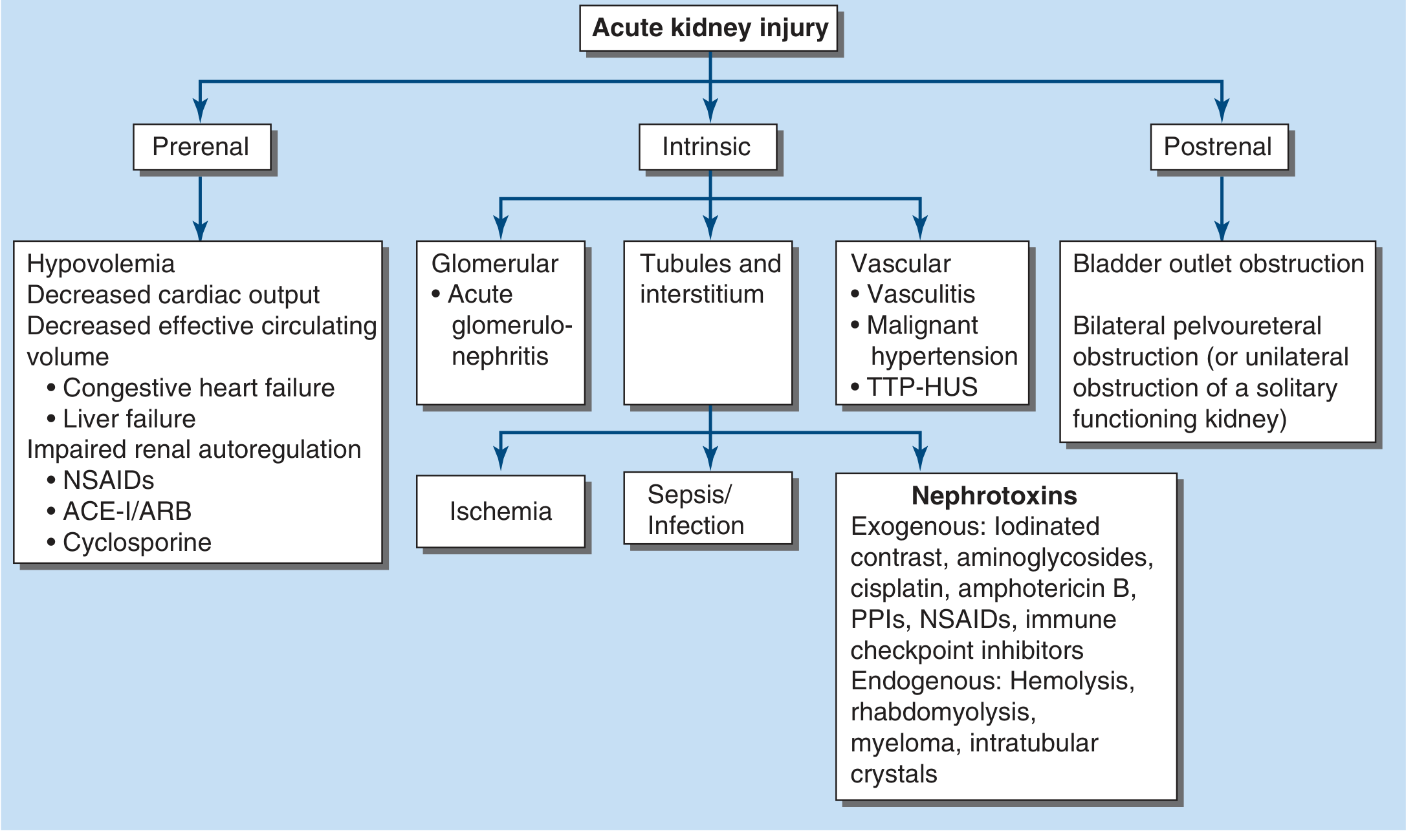
1. Prerenal Azotemia
The most common form - accounts for 40-80% of acute renal failure cases.
Mechanism
Results from inadequate renal plasma flow and intraglomerular hydrostatic pressure to support normal GFR. No intrinsic parenchymal damage occurs - it is rapidly reversible once perfusion is restored.
Causes
- Hypovolemia: GI hemorrhage, diarrhea, vomiting, burns, diuretics
- Decreased cardiac output: Cardiogenic shock, congestive heart failure
- Decreased effective circulating volume: Liver failure (hepatorenal syndrome), sepsis, pancreatitis, peritonitis
- Impaired renal autoregulation: NSAIDs, ACE inhibitors/ARBs, cyclosporine
- Renal vasoconstriction: Severe heart failure, hepatorenal syndrome
Compensatory Response
When perfusion falls, the kidneys activate angiotensin II (efferent arteriole constriction), prostaglandins (afferent arteriole dilation), aldosterone, and ADH/vasopressin to preserve GFR. This is why NSAIDs (block prostaglandins) and ACE-I/ARBs (block angiotensin II) are particularly dangerous in volume-depleted patients - they eliminate both compensatory arms. When mean arterial pressure falls below ~80 mmHg, GFR declines steeply.
Lab Findings (Prerenal vs. ATN)
| Index | Prerenal Azotemia | Oliguric AKI (ATN) |
|---|---|---|
| BUN/Creatinine ratio | >20:1 | 10-15:1 |
| Urine sodium (UNa) | <20 mEq/L | >40 mEq/L |
| Urine osmolality | >500 mOsm/L | <350 mOsm/L |
| FENa | <1% | >2% |
| Urine/plasma creatinine | >40 | <20 |
| Urinalysis (casts) | None or hyaline/granular | Muddy brown granular casts |
Key caveat: FENa can be <1% in early/nonoliguric ATN (e.g., rhabdomyolysis, contrast nephropathy) and in patients with CHF or cirrhosis - so it is not perfectly specific for prerenal azotemia.
Important Notes
- Prolonged prerenal azotemia progresses to acute tubular necrosis (ATN) - ischemic injury with tubular cell death
- SGLT-2 inhibitors (despite lowering intraglomerular pressure) appear to have a protective effect against AKI rather than causing it, per recent data
2. Intrinsic (Renal) Azotemia
Caused by processes acting directly on the renal parenchyma, categorized anatomically:
a. Tubules and Interstitium
- ATN (Acute Tubular Necrosis): Most common intrinsic cause
- Ischemic ATN: Progression from severe prerenal
- Sepsis-associated ATN: AKI complicates >50% of severe sepsis; mechanisms include efferent arteriolar vasodilation, renal vasoconstriction, endothelial damage, microvascular thrombosis, and reactive oxygen species
- Nephrotoxic ATN: Aminoglycosides, cisplatin, amphotericin B, iodinated contrast, NSAIDs, PPIs, immune checkpoint inhibitors
- Endogenous toxins: Hemolysis (hemoglobin), rhabdomyolysis (myoglobin), myeloma (light chains), intratubular crystals (uric acid, oxalate)
- Acute interstitial nephritis (AIN): Drug-induced (penicillins, NSAIDs, PPIs), infection
b. Glomeruli
- Acute glomerulonephritis (post-infectious, IgA nephropathy, RPGN)
- Associated with nephrotic/nephritic syndrome
c. Vasculature
- Vasculitis
- Malignant hypertension
- TTP/HUS (thrombotic microangiopathy)
3. Postrenal Azotemia
Accounts for <5% of acute renal failure but is reversible and must be excluded early.
Requirements for Complete Obstruction
- Obstruction at urethra or bladder outlet (e.g., BPH, stricture), OR
- Bilateral ureteral obstruction, OR
- Unilateral obstruction of a solitary functioning kidney
Causes
- Bladder outlet obstruction (BPH is most common in older men)
- Bilateral ureteral obstruction: Pelvic tumors, retroperitoneal fibrosis, bilateral stones
- Urethral stricture
Mechanism of Azotemia
After obstruction is relieved, there may be a post-obstructive diuresis due to retained solutes and impaired tubular function. Relief of obstruction corrects azotemia.
Diagnosis
Renal ultrasound (hydronephrosis, ureteral dilation) is the first-line test. Note: Early obstruction or ureters encased by tumor/fibrosis may not show dilation.
Azotemia vs. Uremia
| Azotemia | Uremia | |
|---|---|---|
| Definition | Lab finding: elevated BUN/creatinine | Clinical syndrome of renal failure |
| Symptoms | Usually asymptomatic | Nausea, encephalopathy, pericarditis, pruritus |
| GFR threshold | Any reduction in GFR | Typically GFR <15 mL/min |
Uremia develops at variable creatinine levels depending on patient size, age, sex, and underlying disease. Most patients remain asymptomatic until GFR is severely reduced (<15 mL/min).
Evaluation Approach
- History: Volume losses (vomiting, diarrhea, hemorrhage), medications (NSAIDs, ACE-I, diuretics), heart failure, urinary symptoms
- Compare to prior creatinine values - to distinguish acute from chronic
- Urinalysis + microscopy: Casts, protein, blood
- Renal ultrasound: Size (small = chronic), echogenicity, hydronephrosis
- Urinary indices: UNa, FENa, urine osmolality
- Biomarkers: NGAL, KIM-1 (emerging tools for detecting subclinical ATN)
Findings suggesting CKD: anemia, hypocalcemia, hyperphosphatemia, small kidneys with cortical thinning and increased echogenicity on ultrasound, renal osteodystrophy (very late).
Measurement of GFR in Azotemia
- Gold standard: Inulin or iothalamate clearance (not clinically feasible)
- Creatinine clearance (CrCl): 24-hour urine collection; overestimates GFR slightly because creatinine is secreted by proximal tubule
- Serum creatinine: Most widely used surrogate; inversely proportional to GFR. Falls in GFR are reflected proportionally in rising creatinine. Drug dosing adjustments are critical when GFR falls.
- Serum urea (BUN): Underestimates GFR because tubular reabsorption of urea varies with volume status
"Failure to account for GFR reductions in drug dosing can lead to significant morbidity and death from drug toxicities (e.g., digoxin, imipenem)." - Harrison's Principles of Internal Medicine 22E
Sources:
- Harrison's Principles of Internal Medicine, 22E (2025), Chapters 55 and 321
- Goldman-Cecil Medicine International Edition, Chapter on Azotemia
- Morgan & Mikhail's Clinical Anesthesiology, 7e
Treatment for azotemia
Finding Sources
Finding Sources
Searching PubMed
"acute kidney injury" AND treatment management
Reading File
Reading File
Reading File
Reading File
Reading File
I now have all the content needed for a comprehensive treatment overview. Let me compile the full answer.
Treatment of Azotemia
Treatment is directed by the underlying cause - prerenal, intrinsic, or postrenal. The overarching goals are to prevent death, facilitate kidney recovery, and minimize progression to chronic kidney disease (CKD).
Universal Principles (All Types)
Regardless of cause, these measures apply to every patient with azotemia/AKI:
- Optimize hemodynamics - volume resuscitation + vasopressors as needed; maintain mean arterial pressure (MAP) >65 mmHg
- Eliminate nephrotoxins - stop NSAIDs, ACE inhibitors/ARBs, aminoglycosides, contrast agents, chemotherapy, checkpoint inhibitors if at all possible
- Adjust drug doses to current GFR (especially digoxin, imipenem, renally-cleared antibiotics)
- Nutritional support - 20-30 kcal/kg/day; protein 0.8-1.0 g/kg/day (non-dialysis), 1.0-1.5 g/kg/day (dialysis-requiring); prefer enteral route
- Monitor closely - daily serum creatinine, electrolytes, urine output, body weight
1. Prerenal Azotemia
The key principle: restore renal perfusion. Prerenal azotemia is rapidly reversible once hemodynamics are corrected. Importantly, oliguria alone is NOT an indication for fluids - intravascular hypovolemia must be confirmed first.
Fluid Resuscitation
- Match the fluid to the fluid lost:
- Blood loss → packed red blood cells
- Isotonic losses → isotonic crystalloids (preferred over colloids)
- 0.9% saline - most common; avoid in hyperchloremic metabolic acidosis
- Buffered crystalloids (Ringer's lactate, Hartmann's, Plasma-Lyte) - preferred for most patients; avoid if hypernatremic (slightly hypotonic)
- Bicarbonate-containing solutions (e.g., D5W + 150 mEq NaHCO₃) - for metabolic acidosis
- Colloids: Hyperoncotic albumin may be used in liver failure (cirrhosis). Hydroxyethyl starch is no longer available for hospitalized patients due to increased AKI risk.
Optimizing Cardiac Function
- Inotropes (e.g., dobutamine) for cardiogenic causes
- Preload/afterload reduction
- Antiarrhythmics if arrhythmia is compromising output
- Mechanical circulatory support (ventricular assist device) in severe cases
- Consider invasive hemodynamic monitoring (central venous pressure, pulmonary artery catheter) to guide therapy
Hepatorenal Syndrome (Cirrhosis)
A special, challenging prerenal form:
- Exclude spontaneous bacterial peritonitis (SBP) - paracentesis with cell count and culture; albumin infusion reduces AKI risk in SBP treated with antibiotics
- Bridge vasopressor therapy: norepinephrine, terlipressin (vasopressin analogue), or octreotide + midodrine
- Definitive treatment: orthotopic liver transplantation
2. Intrinsic (Renal) Azotemia
No specific pharmacologic agents are available to reverse established ATN or sepsis-associated AKI. Management is primarily supportive while the kidney heals.
General Measures
- Remove the offending nephrotoxin immediately
- Maintain adequate perfusion pressure
- Treat the underlying cause (e.g., antibiotics for sepsis, steroids for AIN, immunosuppression for glomerulonephritis)
Specific Situations
| Cause | Treatment |
|---|---|
| Ischemic ATN / sepsis | Volume resuscitation, vasopressors (norepinephrine), antibiotic therapy, MAP >65 mmHg |
| Rhabdomyolysis | Aggressive IV fluids; consider forced alkaline diuresis (IV sodium bicarbonate to alkalinize urine and prevent myoglobin precipitation) |
| Tumor lysis syndrome | Aggressive IV fluids + allopurinol (xanthine oxidase inhibitor) or rasburicase (uricase - rapidly reduces uric acid) |
| Nephrotoxic drugs (aminoglycosides, cisplatin, contrast) | Discontinue agent; IV fluids pre/post contrast; N-acetylcysteine (limited evidence) |
| Acute interstitial nephritis (AIN) | Withdraw offending drug; corticosteroids (prednisone) in moderate-severe cases |
| Glomerulonephritis (RPGN) | Immunosuppression: corticosteroids ± cyclophosphamide ± rituximab depending on etiology; plasma exchange for anti-GBM disease |
| TTP/HUS | Plasma exchange (TTP); eculizumab (atypical HUS) |
| Contrast nephropathy prevention | IV isotonic saline pre/post procedure; minimize contrast volume |
Prevention of Progression to ATN
- Early recognition and correction of prerenal state before tubular necrosis occurs
- Aggressive volume expansion before major surgery, burns, or trauma
- For sepsis: early antibiotics + rapid hemodynamic stabilization + MAP >65 mmHg; blood glucose target ~140-180 mg/dL (intensive insulin therapy shown to reduce AKI in some studies but increases hypoglycemia risk)
3. Postrenal Azotemia
Treatment = relieve the obstruction as quickly as possible.
| Level of Obstruction | Intervention |
|---|---|
| Urethral / bladder outlet (BPH, stricture) | Transurethral or suprapubic bladder catheter |
| Ureteral obstruction (bilateral or solitary kidney) | Percutaneous nephrostomy tubes OR cystoscopically placed ureteral stents |
| After relief | Urologic evaluation for definitive management of the underlying cause |
Post-Obstructive Diuresis
After obstruction is relieved, a physiologic diuresis occurs (excreting retained solutes/volume) and resolves in days. However, ~5% of patients develop a prolonged salt-wasting syndrome (delayed tubular recovery relative to GFR recovery), requiring IV fluid replacement to maintain blood pressure and prevent hypovolemia.
4. Management of Electrolyte and Metabolic Complications
These arise with any severe azotemia and require active management:
| Problem | Treatment |
|---|---|
| Volume overload | Salt restriction (<1-2 g/day), water restriction (<1 L/day), loop diuretics (if nonoliguric), ultrafiltration/RRT |
| Hyperkalemia | Calcium gluconate 1 g IV (cardiac membrane stabilization if ECG changes); insulin 10-20 units IV + glucose 50% 50 mL; inhaled albuterol (10-20 mg); sodium zirconium cyclosilicate or patiromer (K+ binders); loop diuretics; RRT |
| Metabolic acidosis | Sodium bicarbonate if pH <7.2 or HCO₃⁻ <15 mmol/L; RRT if refractory |
| Hyperphosphatemia | Phosphate dietary restriction; phosphate binders (calcium acetate, sevelamer, aluminum hydroxide) taken with meals |
| Hypocalcemia | Calcium carbonate or calcium gluconate if symptomatic; correct before giving bicarbonate |
| Hyponatremia | Restrict free water; minimize hypotonic IV solutions |
| Hypermagnesemia | Discontinue magnesium-containing antacids |
5. Renal Replacement Therapy (RRT)
Absolute Indications ("AEIOU")
- Acidosis - severe metabolic acidosis refractory to medical management
- Electrolytes - life-threatening hyperkalemia (>6.5 mEq/L or ECG changes) not responding to medical therapy
- Ingestion/Intoxication - dialyzable toxins (e.g., methanol, ethylene glycol, salicylates, lithium)
- Overload - volume overload refractory to diuretics
- Uremia - uremic pericarditis, encephalopathy, neuropathy, or platelet dysfunction (typically BUN >100 mg/dL)
Relative Indications
- Progressive azotemia without uremic manifestations
- Persistent oliguria
Modalities
| Modality | Best For |
|---|---|
| Intermittent Hemodialysis (IHD) | Hemodynamically stable patients; efficient solute clearance |
| Continuous RRT (CRRT) - CVVH, CVVHDF | Hemodynamically unstable/ICU patients; gentler fluid shifts; preferred in sepsis/MODS |
| Slow Low-Efficiency Daily Dialysis (SLEDD) | Hybrid; moderate hemodynamic instability |
| Peritoneal Dialysis | Low-resource settings; patients without vascular access; continuous nature tolerated by hypotensive patients; limited in hypercatabolic states |
Timing of RRT
- The STARRT-AKI trial (NEJM 2020) found no benefit of accelerated (early) vs. standard initiation of RRT, supporting waiting for a clear clinical indication before starting dialysis.
6. Prognosis and Post-AKI Care
- Prerenal azotemia (excluding cardiorenal/hepatorenal syndromes) and postrenal azotemia carry a better prognosis than intrinsic AKI
- The kidney has remarkable self-repair capacity even after dialysis-requiring AKI, when prior renal function was intact
- ~10% of AKI survivors requiring temporary dialysis progress to end-stage kidney disease (ESKD)
- AKI and CKD are interrelated: AKI accelerates CKD progression
- Post-discharge: nephrology follow-up; measure albuminuria (predicts CKD progression risk); aggressive secondary prevention
Sources:
- Harrison's Principles of Internal Medicine, 22E (2025), Chapter 321 - Prevention and Treatment of AKI
- Brenner and Rector's The Kidney, 2-Volume Set, Chapter 29 - Supportive Management of Acute Kidney Injury
- KDIGO Clinical Practice Guidelines for Acute Kidney Injury (Kidney Int Supp 2:1, 2012)
- STARRT-AKI Investigators: Timing of initiation of RRT in AKI. N Engl J Med 383:240, 2020
What is the bridge vasoperssor therapy
Finding Sources
Finding Sources
Searching PubMed
"hepatorenal syndrome" AND terlipressin
Reading File
Reading File
Reading Article
PMID: 38460713
Excellent - I have all the content needed. Here is the full answer:
Bridge Vasopressor Therapy in Hepatorenal Syndrome (HRS)
Background - Why "Bridge" Therapy?
Hepatorenal syndrome (HRS) is a functional kidney injury caused by the hemodynamic derangements of end-stage liver disease and severe portal hypertension. The kidneys themselves are structurally normal - the problem is profoundly reduced renal perfusion driven by:
- Severe splanchnic vasodilation (from portal hypertension)
- Compensatory systemic vasoconstriction that paradoxically impairs renal perfusion
- Activation of the renin-angiotensin-aldosterone system, sympathetic nervous system, and ADH
Since the only definitive treatment is liver transplantation, "bridge" therapy refers to medical interventions used to stabilize or reverse HRS temporarily - keeping the patient alive and preserving renal function while awaiting transplant.
The Three Bridge Vasopressor Regimens
All three regimens work by vasoconstricting the splanchnic circulation, which diverts blood flow back to the systemic and renal circulations, raising MAP and restoring renal perfusion pressure. All are combined with IV albumin (volume expander + oncotic pressure support).
1. Terlipressin + Albumin (First-Line / Most Evidence)
Terlipressin is a synthetic vasopressin analogue that acts primarily on V1 receptors in splanchnic vessels, causing selective splanchnic vasoconstriction.
| Parameter | Detail |
|---|---|
| Dose | 0.5-2.0 mg IV every 4-6 hours (or continuous infusion) |
| Combined with | IV albumin (1.5 g/kg on day 1, then 1 g/kg/day) |
| Effect | Higher reversal rate of HRS vs. placebo |
| Availability | Approved in Europe, some Asian countries; not traditionally available in the US (though FDA approved terlipressin - Terlivaz - for HRS-AKI in 2022) |
Key adverse effects: Respiratory failure/pulmonary edema (most serious - monitor closely), ischemic complications (bowel, skin, digits), hyponatremia.
2. Norepinephrine + Albumin (Alternative When Terlipressin Unavailable)
Norepinephrine is an alpha-1 and alpha-2 adrenergic agonist that causes systemic vasoconstriction, raises MAP, and secondarily improves renal perfusion pressure.
| Parameter | Detail |
|---|---|
| Dose | 0.5-3 mg/hour by continuous IV infusion, titrated to MAP |
| Combined with | IV albumin |
| Setting | Requires ICU monitoring (central line, arterial line) |
| Effectiveness | Comparable to terlipressin in multiple studies |
A 2024 systematic review and meta-analysis (Malik et al., Ann Hepatol) analyzing 9 clinical trials found no significant difference between terlipressin and norepinephrine in reversal of HRS, serum creatinine, urine output, MAP, or mortality - confirming norepinephrine as a safe and effective alternative.
3. Octreotide + Midodrine + Albumin (Least Effective - Outpatient Option)
This combination can be used in patients not requiring ICU-level care, though it is less effective than terlipressin.
| Drug | Class | Mechanism | Route |
|---|---|---|---|
| Midodrine | Alpha-1 agonist | Systemic vasoconstriction; raises MAP | Oral (7.5-12.5 mg three times daily) |
| Octreotide | Somatostatin analogue | Inhibits release of splanchnic vasodilators (glucagon); reduces portal hypertension | Subcutaneous (100-200 mcg three times daily) or IV infusion |
| Albumin | Volume expander | Expands intravascular volume + binds vasodilatory substances | IV |
The advantage of this regimen is that midodrine is oral and octreotide is subcutaneous - it can theoretically be administered outside the ICU, though outcomes are inferior.
Comparison Summary
| Agent | Route | Setting | Effectiveness | Key Concern |
|---|---|---|---|---|
| Terlipressin + albumin | IV bolus/infusion | Ward/ICU | Best evidence | Respiratory failure, ischemia |
| Norepinephrine + albumin | IV infusion | ICU only | Equivalent to terlipressin | Requires ICU monitoring |
| Midodrine + octreotide + albumin | PO/SC | Ward | Least effective | Inferior outcomes |
Other Bridge Strategies (Non-Vasopressor)
- TIPS (Transjugular Intrahepatic Portosystemic Shunt): Reduces portal hypertension directly; can reverse HRS by improving effective circulatory volume. Used in select patients without contraindications (severe hepatic encephalopathy, high MELD score). A 2024 Cochrane systematic review (PMID: 38235907) evaluated TIPS for HRS.
- RRT (Renal Replacement Therapy): Controversial - generally reserved for patients being bridged to transplant when medical therapy fails. Does not treat the underlying hemodynamic cause.
- Extracorporeal Albumin Dialysis (MARS): Experimental; uses albumin dialysate to remove albumin-bound toxins contributing to hemodynamic instability.
Albumin - Why It's Always Co-administered
Albumin is not just a volume expander here. In HRS it:
- Expands intravascular volume (combats the "effective" hypovolemia)
- Binds circulating vasodilatory substances (e.g., bacterial DNA, endotoxins, reactive oxygen species) that drive splanchnic vasodilation
- Reduces systemic inflammation
Sources:
- Goldman-Cecil Medicine International Edition - Hepatorenal Syndrome
- Miller's Anesthesia, 10e - Acute Kidney Injury and Hepatorenal Syndrome
- Harrison's Principles of Internal Medicine, 22E - Prevention and Treatment of AKI
- Malik et al. (2024) - Terlipressin vs. Noradrenaline in HRS: Systematic Review & Meta-Analysis - Ann Hepatol [PMID: 38460713]
What is TIPS for portal hypertension?
Finding Sources
Finding Sources
Searching PubMed
"transjugular intrahepatic portosystemic shunt" AND "portal hypertension"
Reading File
Reading File
Reading Article
PMID: 40180845
Loading Image
Reading Article
PMID: 37141993
I now have comprehensive textbook and current evidence content. Here is the full answer:
TIPS (Transjugular Intrahepatic Portosystemic Shunt) for Portal Hypertension
What Is TIPS?
TIPS is a catheter-based, endovascular procedure in which an interventional radiologist creates an artificial low-resistance channel connecting the portal vein to the hepatic vein through the liver parenchyma. By creating this bypass, blood is diverted away from the high-pressure portal circulation, directly reducing portal hypertension without requiring open surgery.
Procedure - Step by Step
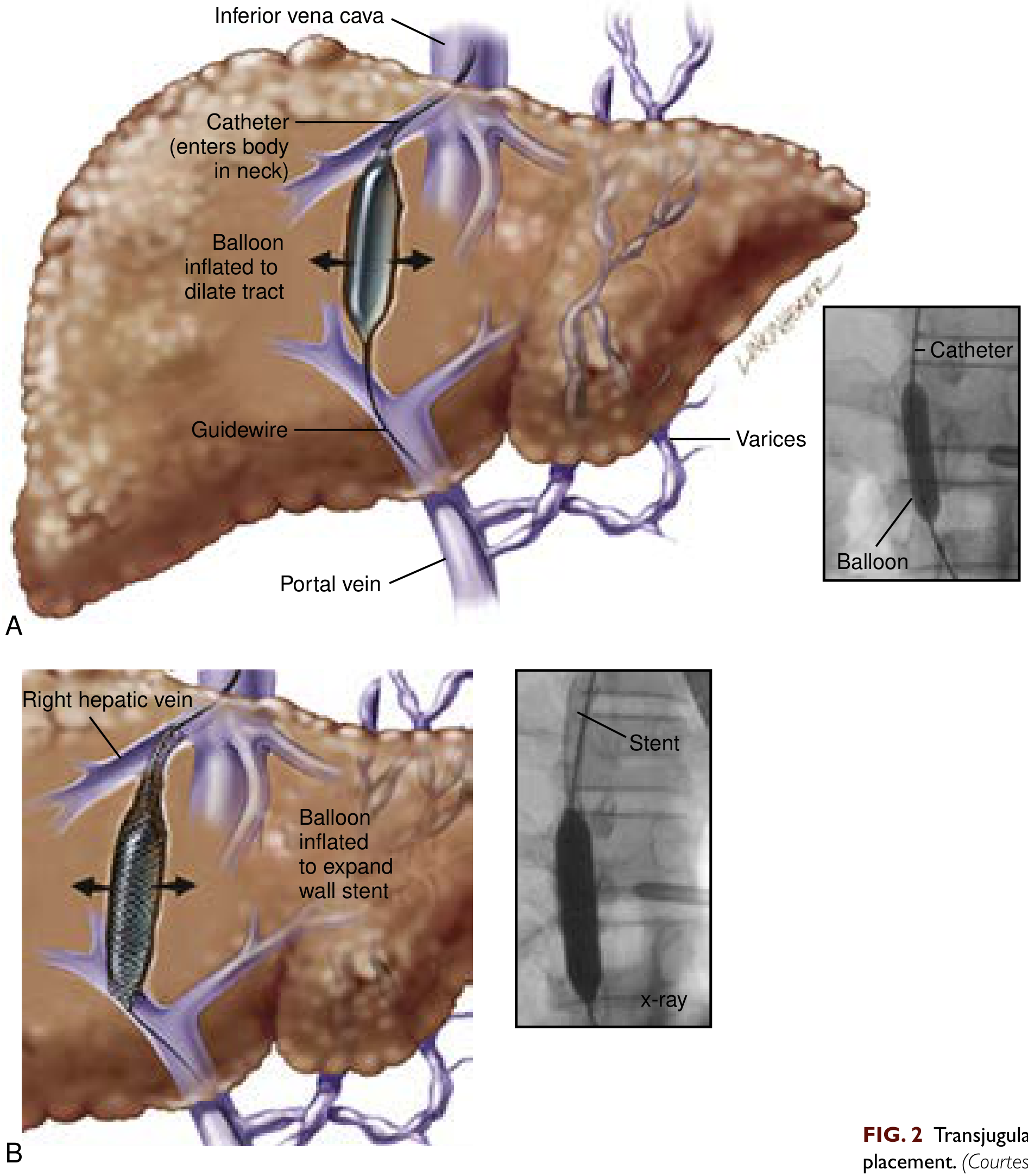
- Access: A catheter is introduced through the right internal jugular vein in the neck
- Navigation: Guidewire and introducer sheath pass through the superior vena cava → inferior vena cava → into a hepatic venous branch
- Portal vein puncture: A needle is passed through the liver parenchyma from the hepatic vein into the portal vein (under fluoroscopic guidance with contrast injection to locate the portal vein)
- Tract dilation: An angioplasty balloon inflates to create a channel along the needle tract
- Stent placement: A polytetrafluoroethylene (PTFE)-covered stent is placed to maintain the shunt. Covered stents have higher patency than older uncovered stents and are now the standard of care.
- Pressure check: The hepatic venous pressure gradient (HVPG) should fall to <12 mmHg after the procedure. Above 12 mmHg, variceal bleeding risk remains elevated.
The procedure can be performed under local anesthesia + sedation or general anesthesia. General anesthesia is preferred in patients with significant ascites or recent variceal hemorrhage (airway protection, ability to maintain supine position).
Why 12 mmHg Matters
| HVPG | Clinical Significance |
|---|---|
| <6 mmHg | Normal |
| 6-10 mmHg | Subclinical portal hypertension |
| ≥10 mmHg | Clinically significant portal hypertension - ascites, varices may develop |
| ≥12 mmHg | Variceal hemorrhage risk threshold |
| ≥20 mmHg | High risk of treatment failure and rebleeding after acute hemorrhage |
TIPS aims to bring HVPG below 12 mmHg, which collapses varices and prevents rebleeding. After a successful TIPS, ascites and esophageal varices may completely resolve over months as portal pressure normalizes.
Indications
Primary / Established Indications
- Refractory variceal hemorrhage - bleeding not controlled by endoscopy (band ligation/sclerotherapy) + pharmacotherapy (vasopressin analogues, beta-blockers)
- Secondary prevention of variceal rebleeding - after a first episode of hemorrhage that failed or is high-risk for endoscopic treatment
- Refractory ascites - ascites not responding to diuretics (spironolactone + furosemide at maximum doses); TIPS reduces need for large-volume paracentesis (LVP)
- Acute variceal hemorrhage in high-risk patients (HVPG >20 mmHg) - early TIPS placement (within 72 hours of endoscopy) reduces rebleeding
Other Indications
- Hepatorenal syndrome (HRS) - as a "bridge" to transplant when vasopressor therapy fails
- Hepatic hydrothorax (refractory pleural effusion from cirrhosis)
- Portal hypertensive gastropathy (refractory to medical therapy)
- Budd-Chiari syndrome (hepatic vein thrombosis causing portal hypertension)
Contraindications
| Absolute | Relative |
|---|---|
| Congestive heart failure | Hepatocellular carcinoma (central tumor near vessels) |
| Severe tricuspid regurgitation | Portal vein thrombosis |
| Severe/moderate pulmonary hypertension | Biliary obstruction |
| Severe hepatic encephalopathy | MELD score ≥18 (higher mortality risk) |
| Sepsis | Severe coagulopathy |
Why cardiac conditions are contraindicated: TIPS diverts portal blood past the liver directly into the right heart, causing an abrupt increase in right-sided venous return. In patients with pre-existing right heart failure, tricuspid regurgitation, or pulmonary hypertension, this can precipitate acute cardiac decompensation.
Stent Size
- Standard stents: 10 mm diameter
- Smaller 8 mm stents have been associated with a lower incidence of hepatic encephalopathy post-TIPS without reducing ascites clearance - a clinically important trade-off in patient selection
Patient Selection - MELD Score
The MELD score (Model for End-Stage Liver Disease) was originally developed to predict 3-month mortality after TIPS. Patients with MELD ≥18 have significantly poorer outcomes. Pre-procedure MELD is a key factor in deciding whether TIPS risk is justified.
Complications
| Complication | Notes |
|---|---|
| Hepatic encephalopathy | Most common; portal blood (containing ammonia and toxins) bypasses hepatic metabolism; occurs in ~20-35% |
| Heart failure | Acute increase in venous return to the right heart |
| Intra-abdominal hemorrhage | ~1-2% incidence; from needle puncture |
| Stent stenosis/thrombosis | Covered PTFE stents have better long-term patency than bare metal; Doppler ultrasound used for surveillance |
| Contrast-induced nephropathy | Relevant in patients with pre-existing renal impairment |
| Hemolytic anemia | From shear forces across the stent |
| Sepsis | Procedure-related infection |
Current Evidence
A landmark 2023 individual patient data meta-analysis (Larrue et al., J Hepatol, PMID: 37141993) analyzing 3,949 patients from 12 controlled studies found:
- 2-year further decompensation rate: 48% with TIPS vs. 63% with standard of care (HR 0.44; 95% CI 0.37-0.54)
- 2-year survival: 71% (TIPS) vs. 63% (standard of care), p = 0.0001
- Benefit was consistent across both indications (refractory ascites and variceal rebleeding prevention)
The EASL 2025 Clinical Practice Guidelines on TIPS [PMID: 40180845] provide the most current comprehensive guidance on all aspects of TIPS in cirrhosis, covering expanding indications, technical advances, and prognostic modelling.
TIPS vs. Alternatives
| Approach | Role |
|---|---|
| TIPS | Best for refractory ascites, secondary variceal prophylaxis; decompresses portal system |
| Endoscopic band ligation | First-line for acute variceal bleeding and primary prevention |
| BRTO (Balloon-occluded retrograde transvenous obliteration) | For gastric varices; preserves portal flow to liver, lower HE risk than TIPS, but can worsen esophageal varices/ascites |
| Surgical portosystemic shunts | Rarely done now; reserved when TIPS is not feasible |
| Liver transplantation | Only definitive treatment for portal hypertension from cirrhosis; TIPS can be a bridge |
Sources:
- Current Surgical Therapy 14e - Transjugular Intrahepatic Portosystemic Shunt
- Miller's Anesthesia, 10e - TIPS Procedure and Anesthesia
- Larrue et al. (2023) - TIPS prevents decompensation and improves survival: IPD meta-analysis, J Hepatol [PMID: 37141993]
- EASL Clinical Practice Guidelines on TIPS (2025), J Hepatol [PMID: 40180845]
Gave me some interesting oncology topics
Here are some fascinating oncology topics worth exploring - spanning basic science, clinical breakthroughs, and emerging frontiers:
🔬 Tumor Biology & Mechanisms
-
Oncogene addiction - How cancers become "addicted" to a single activated oncogene (e.g., EGFR, BCR-ABL) and why this makes them exquisitely sensitive to targeted inhibitors
-
Tumor microenvironment (TME) - The complex ecosystem of cancer cells, immune cells, fibroblasts, and vasculature inside a tumor - and how tumors manipulate it to evade immunity
-
Synthetic lethality - Exploiting two gene defects that are individually tolerable but lethal in combination (e.g., PARP inhibitors in BRCA1/2-mutated cancers)
-
Cancer stem cells - A small subpopulation of cells within tumors with self-renewal capacity, thought to drive recurrence and metastasis
-
Liquid biopsy and circulating tumor DNA (ctDNA) - Detecting and monitoring cancer through blood tests, enabling real-time tracking of tumor evolution and resistance
💊 Treatment & Therapeutics
-
Immune checkpoint inhibitors - PD-1/PD-L1 and CTLA-4 blockade: how they unleash T-cells against tumors, which cancers respond, and the puzzle of why many patients don't
-
CAR-T cell therapy - Engineering a patient's own T-cells to carry a chimeric antigen receptor; remarkable in hematologic malignancies, but hitting walls in solid tumors
-
Antibody-drug conjugates (ADCs) - "Guided missiles" that attach chemotherapy directly to a cancer-targeting antibody; a rapidly expanding field (trastuzumab deruxtecan, sacituzumab govitecan)
-
Bispecific antibodies - Engineered to bind two targets simultaneously - typically a tumor antigen and a T-cell activating receptor (CD3), bringing immune cells directly to tumor cells
-
Tumor-infiltrating lymphocytes (TIL) therapy - Harvesting, expanding, and reinfusing T-cells that have already entered the tumor; FDA approved for melanoma in 2024
🧬 Genetics & Precision Oncology
-
Tumor mutational burden (TMB) - How measuring the total number of mutations in a tumor predicts immunotherapy response, independent of cancer type
-
Microsatellite instability (MSI-H) and mismatch repair deficiency (dMMR) - A pan-cancer biomarker for pembrolizumab response, regardless of tumor origin
-
Clonal hematopoiesis of indeterminate potential (CHIP) - Age-related somatic mutations in blood stem cells; a precursor to hematologic malignancy and a cardiovascular risk factor
-
Epigenetic reprogramming in cancer - How DNA methylation and histone modification patterns are disrupted in cancer, and drugs (DNMT inhibitors, HDAC inhibitors) that target them
⚡ Hot & Emerging Areas
-
Cancer vaccines - Personalized neoantigen mRNA vaccines (Moderna/Merck's mRNA-4157 with pembrolizumab showed strong results in melanoma) - a rapidly evolving space
-
Oncolytic viruses - Genetically engineered viruses that selectively infect and destroy cancer cells while stimulating anti-tumor immunity (T-VEC/talimogene in melanoma)
-
Radioligand therapy - Delivering radiation precisely to cancer cells via targeting molecules (Lutetium-177 PSMA for prostate cancer, Lutetium-177 DOTATATE for neuroendocrine tumors)
-
Resistance mechanisms to targeted therapy - Why cancers almost invariably develop resistance (secondary mutations, bypass signaling, lineage plasticity) and strategies to overcome it
-
Paraneoplastic syndromes - Systemic effects of cancer mediated not by direct invasion but by hormones, antibodies, or immune responses (SIADH, Cushing's, Lambert-Eaton syndrome)
-
Tumor lysis syndrome - The metabolic emergency triggered when rapidly dividing cancers are treated, releasing massive amounts of potassium, phosphate, and uric acid
Just say which topic interests you and I'll give you a deep, textbook-backed breakdown. You can also ask for topics within a specific cancer type (lung, breast, colon, leukemia, etc.) or a particular area like radiation oncology, surgical oncology, or palliative care in cancer.
Explain deep about the tumor infiltrating lymphocytes
Finding Sources
Finding Sources
Searching PubMed
"tumor infiltrating lymphocytes" AND "adoptive cell therapy"
Reading Article
PMID: 39029463
Reading Article
PMID: 38423748
Reading Article
PMID: 37596653
Finding Sources
Searching PubMed
lifileucel melanoma TIL clinical trial FDA
Finding Sources
Finding Sources
Reading File
Reading File
Reading Article
PMID: 42044727
I now have rich content from textbooks, expert consensus guidelines, and the most current real-world data (2026). Let me compile a comprehensive deep-dive.
Tumor-Infiltrating Lymphocytes (TILs): A Deep Dive
1. What Are TILs?
Tumor-infiltrating lymphocytes are immune cells - predominantly T cells - that have naturally migrated from the bloodstream into a tumor. They represent the immune system's own attempt to recognize and destroy cancer. This infiltration is not random: TILs are specifically recruited by chemokines produced in the tumor microenvironment and, critically, many of them carry T-cell receptors (TCRs) that recognize tumor-specific neoantigens - peptides arising from somatic mutations unique to that patient's cancer.
TILs exist on a spectrum:
- CD8+ cytotoxic T cells (CTLs): The primary tumor-killing subset; recognize tumor peptides on MHC class I and directly lyse cancer cells
- CD4+ helper T cells: Support CTL function, activate macrophages, and sustain immune responses
- CD4+ regulatory T cells (Tregs): Immunosuppressive; high Treg density in tumors is associated with worse prognosis
- NK cells, B cells, dendritic cells: Also present in varying proportions
High TIL density in tumors (particularly CD8+ CTLs) is consistently associated with better prognosis in melanoma, colorectal cancer, breast cancer, and others. This observation was the seed from which TIL therapy was developed.
2. Why TILs Fail Naturally - T-Cell Exhaustion & Immune Evasion
Despite TILs being present in tumors, cancer still progresses. The reason is that the tumor microenvironment (TME) actively suppresses and exhausts TILs through multiple mechanisms:
T-Cell Exhaustion
Tumor-infiltrating T cells often display a dysfunctional "exhausted" phenotype, first described in chronic viral infections. Exhausted T cells are characterized by:
- Impaired effector functions (reduced IFN-γ, perforin, granzyme production)
- Progressive loss of proliferative capacity
- Upregulation of multiple inhibitory receptors: PD-1, CTLA-4, LAG-3, TIM-3, TIGIT
This exhaustion is driven by chronic, persistent antigen stimulation by tumor cells over time.
Checkpoint-Mediated Suppression
Tumors exploit T-cell inhibitory (checkpoint) pathways:
| Checkpoint | Ligand on Tumor | Mechanism |
|---|---|---|
| PD-1 | PD-L1 (B7-H1), PD-L2 | Many tumors upregulate PD-L1 (via PDL1 gene amplification or IFN-γ induction); binding PD-1 on TILs blocks effector function |
| CTLA-4 | B7-1, B7-2 (on APCs) | CTLA-4 binds B7 with higher affinity than CD28, competitively blocking T-cell costimulation in lymph nodes |
| LAG-3, TIM-3, TIGIT | Various | Secondary exhaustion markers; co-expression of multiple checkpoint receptors = deeply exhausted T cells |
Other TME Suppression Mechanisms
- TGF-β secretion by tumor cells - directly inhibits lymphocyte and macrophage proliferation and effector function
- Regulatory T cells (Tregs) within tumors suppress anti-tumor CTL responses
- IDO (indoleamine 2,3-dioxygenase) expression - depletes tryptophan, impairing T-cell proliferation
- Myeloid-derived suppressor cells (MDSCs) - produce reactive oxygen species and nitric oxide that impair TIL function
- Hypoxia and nutrient deprivation in the TME further impair T-cell metabolism
The entire rationale for TIL therapy is to extract these cancer-specific T cells from this hostile environment, rescue them ex vivo, expand them massively, and reinfuse them at numbers that overwhelm tumor immune evasion.
3. TIL Therapy: The Process (Manufacturing Protocol)
TIL therapy is a form of adoptive cell therapy (ACT) - transferring immune cells into a patient to treat disease. The manufacturing process is complex, taking approximately 4-6 weeks:
Tumor Resection → Fragment/Digest Tumor →
Initial Expansion (IL-2) → REP (Rapid Expansion Protocol) →
Patient Lymphodepletion → TIL Infusion → IL-2 Support
Step 1 - Tumor Harvest (Surgical)
- A tumor fragment (≥1.5 cm is typically required) is surgically resected
- The tumor is dissected into small fragments or enzymatically digested into single-cell suspensions
- TILs are extracted from the tumor tissue
Step 2 - Initial Expansion ("Pre-REP")
- Extracted TIL fragments are cultured in media containing high-dose IL-2 (interleukin-2)
- IL-2 drives T-cell proliferation and survival
- Duration: ~2 weeks; produces a preliminary TIL population from the tumor fragments
Step 3 - Rapid Expansion Protocol (REP)
- TILs undergo a second, more aggressive expansion using:
- Anti-CD3 antibody (OKT3) - provides non-specific TCR stimulation
- High-dose IL-2
- Irradiated feeder cells (allogeneic PBMCs) - provide co-stimulatory signals
- REP expands TILs by 1,000-fold or more over ~2 weeks
- Final product: 50 billion - 150 billion TIL cells
Step 4 - Non-Myeloablative Lymphodepletion (Preparative Regimen)
Before TIL infusion, the patient receives lymphodepleting chemotherapy - typically:
- Cyclophosphamide 60 mg/kg/day × 2 days
- Fludarabine 25 mg/m²/day × 5 days
Why lymphodepletion is critical:
- Eliminates endogenous lymphocytes that would compete with TILs for homeostatic cytokines (IL-7, IL-15, IL-2)
- Removes immunosuppressive Tregs from circulation
- Creates "homeostatic space" - a cytokine-rich environment that drives rapid TIL expansion after infusion
- Eliminates MDSCs and other immunosuppressive cells temporarily
This step is considered essential - TIL therapy without prior lymphodepletion has significantly lower efficacy.
Step 5 - TIL Infusion
- Billions of TILs are infused intravenously (single infusion)
- TILs home to tumor sites via adhesion molecules and chemokine gradients
Step 6 - High-Dose IL-2 Support
- After TIL infusion, patients receive high-dose IL-2 (aldesleukin, 600,000-720,000 IU/kg IV every 8 hours, up to 5 doses)
- IL-2 supports the survival and expansion of infused TILs in vivo
- Major toxicity: capillary leak syndrome (hypotension, pulmonary edema, fever) - requires ICU-level monitoring; limits use to fit patients
4. FDA Approval - Lifileucel (Amtagvi)
In February 2024, the FDA approved lifileucel (Amtagvi, Iovance Biotherapeutics) - the first TIL cell therapy product ever approved, and the first FDA-approved cellular therapy for a solid tumor.
Indication
Unresectable or metastatic melanoma in adults who have received:
- A prior anti-PD-1 therapy (pembrolizumab/nivolumab)
- If BRAF V600-mutant: also a prior BRAF inhibitor ± MEK inhibitor
Pivotal Trial - C-144-01 (KEYNOTE-119 substudy)
- Single-arm, multicenter Phase 2 trial
- Overall Response Rate (ORR): 31.5% in heavily pre-treated patients
- Complete response rate: ~5% (potentially durable/curative)
- Median duration of response: not reached in responders at time of analysis
- Population: patients who had failed checkpoint inhibitors and often targeted therapies
Real-World Data (2026)
A 2026 real-world study (Desai et al., PMID: 42044727) from a single-center TIL program (45 melanoma patients) found:
- Only 51% of referred patients completed TIL infusion (barriers: small tumor size <1.5 cm, comorbidities, rapid disease progression, out-of-spec product)
- 1-year OS: 79.2% in patients who successfully received TIL therapy vs. 42% in those who did not (p = 0.007)
- Median time from referral to TIL infusion was 128 days (reduced to 96 days with workflow optimization)
- Highlights that real-world outcomes may be lower than clinical trial ORR due to patient selection issues
5. TIL Density as a Prognostic Biomarker
Independent of TIL therapy, the density and spatial distribution of TILs within a tumor has prognostic value across many cancer types:
| TIL Pattern | Description | Prognostic Significance |
|---|---|---|
| "Hot" (inflamed) | High TIL density throughout tumor | Best prognosis; most likely to respond to checkpoint inhibitors |
| "Excluded" | TILs present at tumor margin but excluded from core | Intermediate; TGF-β often drives this pattern |
| "Cold" (desert) | Few or no TILs | Worst prognosis; resistant to immunotherapy |
The Immunoscore - a standardized scoring system based on CD3+ and CD8+ T-cell densities at the tumor core and invasive margin - has been validated as a prognostic tool in colorectal cancer and is under evaluation in other cancers.
6. Challenges and Limitations
| Challenge | Detail |
|---|---|
| Manufacturing complexity | 4-6 week process at specialized centers; tumor must be surgically accessible (≥1.5 cm) |
| Patient fitness requirement | Lymphodepletion + high-dose IL-2 require good performance status; not suitable for frail patients |
| IL-2 toxicity | Capillary leak syndrome requires ICU monitoring; limits broad use |
| Out-of-spec products | ~28.5% of TIL products in one real-world series failed quality standards |
| Limited efficacy in solid tumors beyond melanoma | Immunosuppressive TME, poor TIL infiltration, antigen heterogeneity |
| Cost and access | Highly centralized manufacturing; expensive and logistically complex |
| Time pressure | Rapidly progressing melanoma may not allow 4-6 weeks for manufacturing |
7. Next-Generation TIL Approaches
Ongoing research is addressing current limitations:
- Selected/enriched TILs: Selecting for tumor-reactive T cells (e.g., by PD-1 selection or neoantigen reactivity screening) rather than bulk expansion - higher potency with fewer cells
- Shorter manufacturing ("Fresh TIL"): Bypassing the REP step to reduce time to infusion
- Reduced-dose IL-2 regimens: Using IL-2 variants (e.g., bempegaldesleukin) or other cytokines (IL-15, IL-21) to support TIL survival with less toxicity
- TIL + checkpoint inhibitor combinations: Combining TIL infusion with anti-PD-1 to prevent re-exhaustion
- Genetically engineered TILs: Inserting transgenes into TILs (e.g., knockout of PD-1, insertion of cytokine transgenes) to make them exhaustion-resistant
- Allogeneic TIL therapy ("off-the-shelf"): Manufacturing TILs from healthy donors to reduce lead time and cost
- Expansion to other solid tumors: Active trials in cervical cancer, NSCLC, head and neck, colorectal cancer
8. TIL Therapy vs. Other Adoptive Cell Therapies
| Feature | TIL Therapy | CAR-T | TCR-T |
|---|---|---|---|
| Antigen targeting | Polyclonal - multiple tumor antigens | Single antigen (e.g., CD19, BCMA) | Single specific neoantigen/tumor antigen |
| Source | Tumor-derived (already tumor-reactive) | Peripheral blood (engineered) | Peripheral blood (engineered) |
| Applicability | Solid tumors (primarily) | Hematologic malignancies (mainly) | Solid tumors (experimental) |
| Manufacturing | 4-6 weeks; complex | 2-4 weeks; complex | Experimental |
| FDA approval | Melanoma (2024) | Hematologic malignancies (B-ALL, DLBCL, MM, MCL) | None yet |
| Key limitation | IL-2 toxicity, surgical requirement | CRS, neurotoxicity; poor solid tumor penetration | HLA restriction; antigen loss |
The polyclonal nature of TILs - attacking multiple tumor antigens simultaneously - is a major theoretical advantage over CAR-T (which targets only one antigen and risks antigen escape), particularly in the heterogeneous tumor antigen landscape of solid tumors.
Sources:
- Cellular and Molecular Immunology (Abbas) - Molecules and Cells That Inhibit Immune Responses to Tumors
- Schwartz's Principles of Surgery, 11e - Immunotherapy in Cancer
- Sarnaik et al. (2024) - "Tumor-Infiltrating Lymphocytes: A New Hope", Cancer Cell [PMID: 39029463]
- Betof Warner et al. (2024) - Expert Consensus Guidelines on TIL Cell Therapy, J Immunother Cancer [PMID: 38423748]
- Desai et al. (2026) - Real-World Outcomes with Commercial Lifileucel, Transplant Cell Ther [PMID: 42044727]
- Zhang et al. (2023) - Challenges and New Technologies in Adoptive Cell Therapy, J Hematol Oncol [PMID: 37596653]
This is a shared conversation. Sign in to Orris to start your own chat.