Explain the laboratory approach to diagnose a male child with repeated joint bleeding. Pathology very briefly
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Finding Sources
I have all the information needed. Let me now compile the complete, well-organized answer:
Laboratory Approach to Diagnose a Male Child with Repeated Joint Bleeding (Hemarthrosis)
Brief Pathology
Repeated hemarthrosis in a male child indicates a coagulation factor deficiency of the intrinsic (contact activation) pathway. The X-linked recessive inheritance pattern in a male child points overwhelmingly to:
- Hemophilia A - Factor VIII deficiency (incidence: 1 in 5,000 live male births)
- Hemophilia B (Christmas disease) - Factor IX deficiency (incidence: 1 in 30,000 males)
Both disorders leave the extrinsic (tissue factor) pathway and platelet plug formation intact, which explains why small cuts stop bleeding normally but deep tissue and joint bleeds occur. Recurrent hemarthrosis triggers proliferative synovitis and cartilage degradation, eventually leading to erosive arthropathy. Almost all patients with factor levels <1% of normal experience spontaneous hemarthroses involving large joints (knees, elbows, ankles).
Step-by-Step Laboratory Approach
Step 1: Initial Screening Tests
| Test | Expected Result in Hemophilia | Rationale |
|---|---|---|
| aPTT (Activated Partial Thromboplastin Time) | Prolonged | Tests intrinsic + common pathway; factors VIII and IX are in this pathway |
| PT (Prothrombin Time) | Normal | Tests extrinsic pathway only (Factor VII, X, V, II, fibrinogen) |
| Thrombin Time (TT) | Normal | Normal TT helps exclude heparin contamination as a cause of prolonged aPTT |
| Platelet count | Normal | Bleeding here is not platelet-type; rules out thrombocytopenia |
| Bleeding time / PFA-100 | Normal | Primary hemostasis is intact; platelet plug forms normally |
| Complete Blood Count | May show anemia if bleeds are significant | Baseline assessment |
Key pattern: Isolated prolonged aPTT with normal PT and normal platelet count = intrinsic pathway factor deficiency.
Step 2: aPTT Mixing Studies
A fresh 1:1 mix of patient plasma + normal pooled plasma is tested:
- Correction of aPTT → suggests a factor deficiency (missing factor is supplied by normal plasma)
- No correction → suggests an inhibitor (e.g., lupus anticoagulant or an acquired factor inhibitor)
Important nuance: Factor VIII inhibitors are time-dependent. An immediate mix may correct, but a 1-hour incubated mix will show prolongation. Always run both immediate and incubated mixing studies.
Step 3: Specific Factor Assays (Confirmatory)
In a male child, the following intrinsic pathway factors are measured:
| Factor | Deficiency = Diagnosis |
|---|---|
| Factor VIII activity | Low + normal IX & XI = Hemophilia A |
| Factor IX activity | Low + normal VIII & XI = Hemophilia B |
| Factor XI activity | Low = Hemophilia C (autosomal recessive, usually milder) |
Note: Factor XII, prekallikrein, and HMWK deficiencies also prolong aPTT but do not cause clinical bleeding - they are excluded by clinical context.
Step 4: Severity Classification (Factor VIII or IX level)
| Factor Activity | Severity | Clinical Manifestations |
|---|---|---|
| < 1 IU/dL (<1%) | Severe | Spontaneous hemarthroses, muscle bleeds |
| 1-5 IU/dL (1-5%) | Moderate | Bleeding after minor trauma |
| >5-40 IU/dL (5-40%) | Mild | Bleeding only after surgery or significant trauma |
Step 5: Distinguish from Von Willebrand Disease (vWD)
vWD (especially type 3 and type 2N) can also present with low factor VIII and prolonged aPTT:
| Test | Hemophilia A | vWD Type 2N |
|---|---|---|
| Factor VIII activity | Low | Low |
| VWF antigen (VWF:Ag) | Normal | Low or normal |
| VWF ristocetin cofactor (VWF:RCo) | Normal | Reduced |
| VWF:VIII binding assay | Normal | Reduced (hallmark of type 2N) |
| Inheritance | X-linked (males affected) | Autosomal recessive |
The VWF:VIII binding assay distinguishes type 2N vWD from hemophilia A but is only available at specialized labs. In a male child with no female relatives affected, X-linked hemophilia A is far more likely than type 2N vWD.
Step 6: One-Stage vs. Two-Stage / Chromogenic Assay
- The standard one-stage clot-based aPTT assay is used most commonly.
- A two-stage or chromogenic factor VIII assay may give discrepant (lower) results in mild discrepant hemophilia A, where up to 40% of mild cases are missed by the one-stage assay alone.
- The World Federation of Hemophilia recommends performing both at initial diagnosis.
Step 7: Inhibitor Detection (if treatment fails)
If a diagnosed hemophiliac fails to respond to factor replacement:
- Repeat aPTT: prolonged
- Mixing study: no correction (inhibitor pattern)
- Bethesda assay: quantifies inhibitor titer in Bethesda Units (BU/mL)
- < 5 BU/mL = low-titer inhibitor (may still respond to high-dose factor)
- ≥ 5 BU/mL = high-titer inhibitor (requires bypassing agents: rFVIIa or aPCC)
Step 8: Genetic / Molecular Testing
- Identifies the specific F8 or F9 gene mutation
- Useful for carrier detection in female relatives and prenatal diagnosis
- A family history may be absent in up to 30% of infant males with hemophilia A (spontaneous mutations)
Diagnostic Algorithm Summary
Male child + recurrent hemarthrosis
↓
Screening: CBC, PT, aPTT, TT, Bleeding time
↓
Isolated ↑ aPTT, normal PT, normal platelets
↓
aPTT Mixing Study
↙ ↘
Corrects No correction
(Deficiency) (Inhibitor → Bethesda assay)
↓
Measure Factor VIII, IX, XI activities
↙ ↘
Low FVIII Low FIX
(normal IX/XI) (normal VIII/XI)
↓ ↓
Hemophilia A Hemophilia B
↓
Severity classification
+ vWD workup if needed
+ Molecular testing
Sources:
- Tietz Textbook of Laboratory Medicine, 7th Edition - Inherited Coagulation Disorders, Factor VIII & Factor IX Deficiency sections (pp. 3122-3124)
- Goldman-Cecil Medicine - Hemophilia and Arthritis chapter (p. Hemophilia section)
Explain the laboratory approach to diagnose a male child with repeated joint bleeding. Pathology briefly
This is the same question as before — I already have the full textbook content in context. Here is the complete answer with the pathology section expanded slightly per your request:
Laboratory Diagnosis of Repeated Joint Bleeding (Hemarthrosis) in a Male Child
Pathology (Brief)
Repeated hemarthrosis in a male child is the hallmark of an X-linked recessive coagulation factor deficiency, most commonly:
- Hemophilia A - Factor VIII deficiency (1 in 5,000 male births)
- Hemophilia B (Christmas disease) - Factor IX deficiency (1 in 30,000 males)
Pathophysiology of bleeding:
Both factors VIII and IX are essential components of the intrinsic (contact activation) pathway of coagulation. Factor IXa and Factor VIIIa together form the tenase complex, which activates Factor X. Without this complex, the coagulation cascade cannot generate sufficient thrombin to form a stable fibrin clot in deep tissues and joints. Platelet plug formation (primary hemostasis) remains intact, so superficial cuts stop bleeding normally - only deep tissue, muscle, and joint bleeds occur.
Pathophysiology of joint damage:
Recurrent bleeding into a joint causes:
- Acute synovitis - blood in the joint space triggers an inflammatory response
- Iron deposition - repeated bleeds deposit hemosiderin in the synovium, stimulating synoviocyte proliferation
- Chronic proliferative synovitis - the hypertrophied synovium becomes a source of further bleeding
- Cartilage degradation - proteolytic enzymes from activated synoviocytes destroy articular cartilage
- End-stage hemophilic arthropathy - bone enlargement, crepitus, muscle atrophy, joint contractures, and complete loss of joint space
Large joints are preferentially affected: knees > elbows > ankles. Almost all patients with factor levels < 1% of normal experience spontaneous hemarthroses.
Laboratory Approach: Step by Step
Step 1 - Initial Screening Tests
These tests are performed first to characterize the type of hemostatic defect:
| Test | Result in Hemophilia | Why |
|---|---|---|
| aPTT | Prolonged | Intrinsic pathway is defective (Factors VIII or IX are missing) |
| PT | Normal | Extrinsic pathway (Factor VII) and common pathway are intact |
| Thrombin Time (TT) | Normal | Fibrinogen is normal; also excludes heparin contamination |
| Platelet count | Normal | This is not a platelet disorder |
| Bleeding time / PFA-100 | Normal | Primary hemostasis (platelet plug) is intact |
| CBC | May show anemia if bleeds are large | Baseline assessment |
Diagnostic pattern: Isolated prolonged aPTT + normal PT + normal platelet count + normal bleeding time = intrinsic pathway factor deficiency
Step 2 - aPTT Mixing Study
Fresh patient plasma is mixed 1:1 with normal pooled plasma and the aPTT is repeated:
| Mixing Study Result | Interpretation |
|---|---|
| aPTT corrects to normal | Factor deficiency - normal plasma supplied the missing factor |
| aPTT does not correct | Inhibitor present (lupus anticoagulant, or acquired factor inhibitor) |
Important: Factor VIII inhibitors are time-dependent. An immediate mix may falsely correct. Always run an incubated (1-hour at 37°C) mixing study alongside the immediate mix to detect time-dependent inhibitors.
Step 3 - Specific Factor Assays (Confirmatory)
Once a factor deficiency pattern is established, measure individual intrinsic pathway factors. In a male child, the key assays are:
| Factor Assay | Result | Diagnosis |
|---|---|---|
| Factor VIII activity low, IX and XI normal | ↓ VIII | Hemophilia A |
| Factor IX activity low, VIII and XI normal | ↓ IX | Hemophilia B |
| Factor XI activity low | ↓ XI | Hemophilia C (autosomal, milder) |
Contact factors (XII, prekallikrein, HMWK) also prolong aPTT but do not cause clinical bleeding - they are excluded by the clinical context of hemarthrosis.
Step 4 - Severity Classification
Based on the factor activity level:
| Factor Activity | Severity | Clinical Pattern |
|---|---|---|
| < 1 IU/dL (< 1%) | Severe | Spontaneous joint and muscle bleeds |
| 1 - 5 IU/dL (1-5%) | Moderate | Bleeds after minor trauma |
| > 5 - 40 IU/dL (5-40%) | Mild | Bleeds only after surgery or significant injury |
Step 5 - Exclude Von Willebrand Disease (vWD)
vWD (especially type 2N) can mimic Hemophilia A because VWF normally protects Factor VIII from degradation. When VWF has decreased affinity for Factor VIII (type 2N), Factor VIII levels fall and aPTT is prolonged:
| Test | Hemophilia A | vWD Type 2N |
|---|---|---|
| Factor VIII | Low | Low |
| VWF antigen | Normal | Low or normal |
| VWF ristocetin cofactor | Normal | Reduced |
| VWF:VIII binding assay | Normal | Reduced (diagnostic) |
| Inheritance | X-linked (males >> females) | Autosomal recessive |
In a male child with no females affected, Hemophilia A is far more likely. However, in females or where family history is atypical, vWD workup is essential before rendering a final diagnosis.
Step 6 - One-Stage vs. Chromogenic Assay
- The one-stage clot-based aPTT assay is standard.
- A chromogenic (two-stage) Factor VIII assay may give discrepant (lower) results in mild discrepant Hemophilia A - up to 40% of mild cases can be missed by one-stage assay alone.
- The World Federation of Hemophilia recommends performing both at the time of initial diagnosis.
Step 7 - Inhibitor Detection (if factor replacement fails)
When a known hemophiliac does not respond to factor replacement therapy:
- aPTT remains prolonged despite treatment
- Mixing study: no correction (inhibitor present)
- Bethesda Assay: quantifies inhibitor titer in Bethesda Units (BU/mL)
| Bethesda Titer | Classification | Management |
|---|---|---|
| < 5 BU/mL | Low-titer inhibitor | High-dose factor replacement may work |
| ≥ 5 BU/mL | High-titer inhibitor | Bypassing agents required (rFVIIa or aPCC) |
Inhibitors occur in 10-25% of Hemophilia A patients (usually severe disease) and only 1-3% of Hemophilia B patients. An anamnestic response (rising inhibitor titer 4-7 days after re-exposure to factor) must be anticipated.
Step 8 - Molecular / Genetic Testing
- Identifies the specific F8 or F9 gene mutation
- Used for carrier detection in female relatives
- Used for prenatal diagnosis in subsequent pregnancies
- Important note: up to 30% of Hemophilia A cases arise from de novo spontaneous mutations - absence of family history does not exclude the diagnosis
Summary Diagnostic Algorithm
Male child + recurrent hemarthrosis
↓
CBC, PT, aPTT, TT, Bleeding time
↓
Isolated prolonged aPTT
Normal PT, Normal platelets, Normal bleeding time
↓
aPTT Mixing Study
↙ ↘
Corrects No correction
(Deficiency) (Inhibitor)
↓ ↓
Measure F VIII, F IX, Bethesda Assay
F XI activities (quantify inhibitor)
↙ ↘
Low F VIII Low F IX
= Hemophilia A = Hemophilia B
↓
Severity by activity level
+ vWD workup if needed
+ One-stage AND chromogenic assay
+ Molecular testing
Sources:
- Tietz Textbook of Laboratory Medicine, 7th Ed. - Inherited Coagulation Disorders; Factor VIII Deficiency (Hemophilia A); Factor IX Deficiency (Hemophilia B) (pp. 3122-3124)
- Goldman-Cecil Medicine - Hemophilia and associated arthropathy
Explain the correlation between cigarette smoking and the development of squamous cell carcinoma of the lung. Pathology briefly
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed for a complete, image-supported answer. Here it is:
Cigarette Smoking and Squamous Cell Carcinoma of the Lung
Pathology (Brief)
Gross Pathology
Squamous cell carcinoma (SCC) is a centrally located tumor, classically arising from the bronchial epithelium of segmental or lobar bronchi near the hilum. It forms a firm, gray-white intraluminal or peribronchial mass that may cause bronchial obstruction, leading to post-obstructive pneumonia or atelectasis distally. Central necrosis and cavitation are common due to outgrowing blood supply.
Histopathology
The defining microscopic features of SCC are:
- Intercellular bridges (desmosomes between squamous cells)
- Individual cell keratinization (bright pink cytoplasm)
- Keratin pearls - concentric whorls of keratinizing squamous cells (hallmark of well-differentiated SCC)
- Nuclear pleomorphism, hyperchromasia, and mitotic figures
- Ranges from well-differentiated (abundant keratin) to poorly differentiated (squamous features minimal or absent)
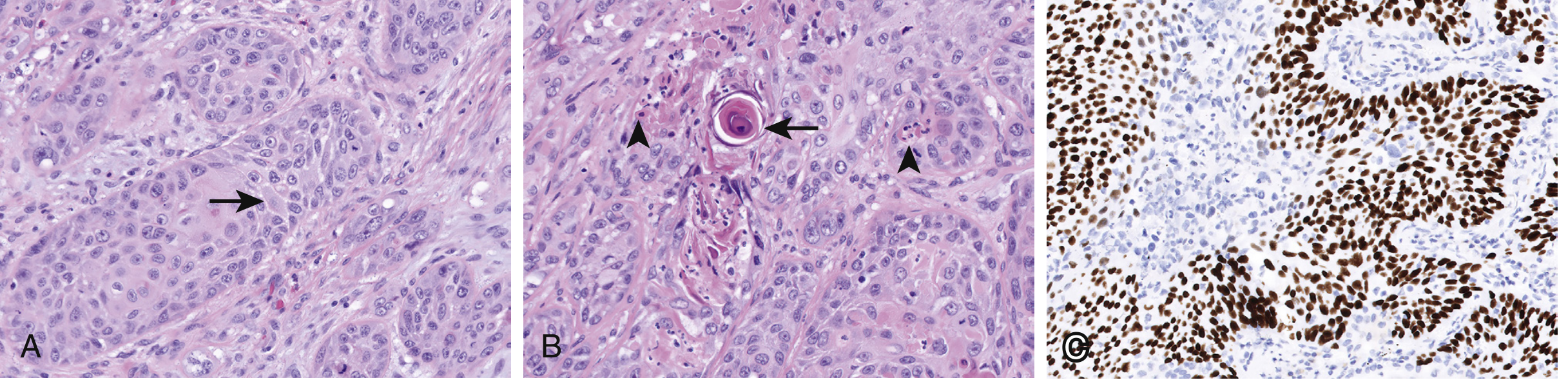
Immunohistochemistry
- p63 / p40 - nuclear positive (hallmark of squamous differentiation)
- TTF-1 and Napsin A - negative (distinguishes from adenocarcinoma)
- CK5/6 - positive
The Smoking-SCC Correlation: Step by Step
1. Epidemiology
- ~80% of all lung cancers occur in active smokers or recent ex-smokers
- SCC is the lung cancer subtype most tightly linked to tobacco - it is rare in never-smokers
- Risk is 60 times greater in heavy smokers (2 packs/day for 20 years) vs. nonsmokers
- Incidence correlates nearly linearly with pack-years
- SCC accounts for approximately 25% of all lung cancers in the United States
- Historically up to two-thirds of SCC presented as central hilar tumors; peripheral SCC has increased with the shift to filtered cigarettes
2. Carcinogens in Cigarette Smoke
Tobacco smoke contains >60 known carcinogens acting through two main chemical classes:
| Carcinogen Class | Examples | Mechanism |
|---|---|---|
| Polycyclic aromatic hydrocarbons (PAHs) | Benzo[a]pyrene | Require P-450 activation → form bulky DNA adducts → G→T transversions |
| Nitrosamines | NNK, NNN | Alkylate DNA → mutations in TP53, KRAS |
| Benzene, formaldehyde, metals (Cr, Ni, As) | Various | DNA strand breaks, cross-links |
These are procarcinogens that require metabolic activation by the cytochrome P-450 monooxygenase system (especially CYP1A1, CYP1B1). Individuals with P-450 polymorphisms that increase activation capacity have a greater risk of lung cancer from the same exposure.
3. "Field Cancerization" Effect
Cigarette smoke mutagenizes the entire respiratory mucosa simultaneously, not just one cell. This means:
- Chromosome deletions at 3p, 9p (CDKN2A locus), and 17p (TP53 locus) are detectable in histologically normal bronchial epithelium of smokers - long before any visible tumor
- This "field effect" explains why multiple synchronous or metachronous lung cancers can arise, and why former smokers retain elevated risk even after cessation
- Genetic changes can persist for many years in bronchial epithelium after quitting smoking
4. Stepwise Progression: The Metaplasia-Dysplasia-Carcinoma Sequence
This is the best-characterized carcinogenesis sequence in all of lung cancer. There is a linear correlation between cigarette smoke intensity and the stage of epithelial change:
Normal pseudostratified
ciliated columnar epithelium
↓ (smoke → chronic irritation)
Goblet cell hyperplasia
↓
Basal cell (reserve cell) hyperplasia
↓
Squamous metaplasia
(normal columnar epithelium replaced by stratified squamous epithelium)
↓
Squamous dysplasia
(disordered squamous epithelium: loss of polarity, nuclear hyperchromasia,
pleomorphism, mitoses, increased N:C ratio)
↓ (Mild → Moderate → Severe dysplasia)
Carcinoma in situ (CIS)
(full-thickness cytologic atypia; basement membrane intact)
↓
Invasive Squamous Cell Carcinoma
(basement membrane breached; invasion into stroma and lymphovascular channels)
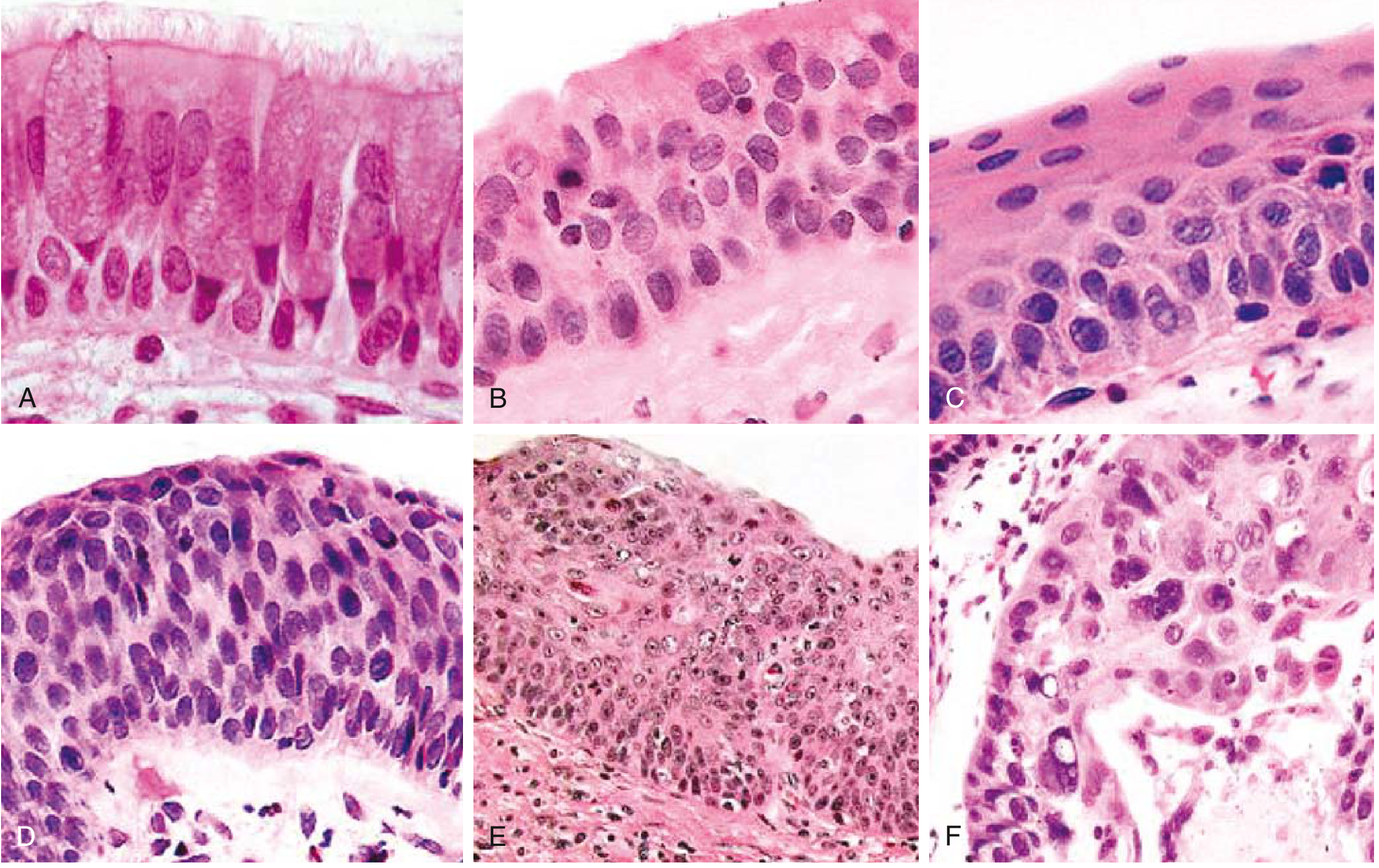
5. Molecular/Genetic Events (SCC-Specific)
SCC harbors diverse chromosomal deletions targeting tumor suppressor loci rather than the targetable tyrosine kinase mutations seen in adenocarcinoma:
| Genetic Event | Frequency | Significance |
|---|---|---|
| TP53 mutation | 65-90% | Earliest detectable event - p53 overexpression in 10-50% of squamous dysplasias and 60-90% of CIS |
| CDKN2A (p16) deletion/mutation at 9p | 65% | Loss of cell cycle brake at G1/S checkpoint; allows uncontrolled proliferation |
| 3p deletion | Early, nearly universal | Loss of multiple tumor suppressor loci (including FHIT, RASSF1); detected even in normal epithelium of smokers |
| 17p (TP53) deletion | Early event | Genomic instability |
| PIK3CA mutation | 30% | Activates PI3K/Akt survival signaling |
| SOX2 amplification | 15% | Squamous lineage transcription factor; promotes squamous phenotype |
| FGFR1 amplification | ~20% | Fibroblast growth factor receptor; potential therapeutic target |
| CCND1 amplification | 15% | Cyclin D1 overexpression; drives G1 → S cell cycle progression |
Key contrast: Unlike adenocarcinoma (which has actionable EGFR, ALK, ROS1 mutations), SCC mutations are currently not actionable for targeted therapy. Treatment relies on immunotherapy (PD-L1 targeting) and chemotherapy.
6. Why Squamous Cell and Not Adenocarcinoma?
| Feature | Squamous Cell Carcinoma | Adenocarcinoma |
|---|---|---|
| Smoking association | Very strong (rare in never-smokers) | Moderate (most common in never-smokers) |
| Location | Central (hilar bronchi) | Peripheral (alveoli/small airways) |
| Cell of origin | Bronchial basal/reserve cells → squamous metaplasia | Bronchioalveolar stem cells / club cells |
| Key mutations | TP53, CDKN2A, PIK3CA, FGFR1 | EGFR, KRAS, ALK, ROS1 |
| Precursor lesion | Squamous metaplasia → dysplasia → CIS | Atypical adenomatous hyperplasia → AIS |
The reason smoke preferentially causes squamous transformation in central airways is the direct, concentrated exposure of large bronchi to inhaled carcinogens, combined with the capacity of basal/reserve cells in these airways to undergo squamous metaplasia as a protective response to chronic irritation - a response that, when sustained and genetically destabilized, progresses to malignancy.
7. Other Risk Modifiers
- Secondhand smoke: Contains the same carcinogens; ~3,000 nonsmoker lung cancer deaths/year in the US
- Industrial co-carcinogens: Asbestos (especially with smoking - synergistic, not additive), arsenic, chromium, nickel, uranium - all amplify SCC risk
- Radon gas: Radioactive decay produces alpha-particle-emitting isotopes that cause direct DNA strand breaks in bronchial epithelium
- Genetic susceptibility: P-450 polymorphisms and DNA repair gene variants (mutagen sensitivity genotype) confer >10-fold higher risk in smokers vs. controls with the same exposure
- Cessation: Reduces risk over time but never returns to baseline due to persisting epigenetic and genetic changes in the field-mutagenized epithelium
Sources:
- Robbins & Cotran Pathologic Basis of Disease - Etiology and Pathogenesis of Lung Cancer; Acquired Mutations (pp. 668-670)
- Robbins & Kumar Basic Pathology - Morphology and Carcinogenesis sequence (pp. 486-488)
- Murray & Nadel's Textbook of Respiratory Medicine - Squamous Cell Carcinoma (p. 562)
This is a shared conversation. Sign in to Orris to start your own chat.