Explain me thalassemia from basic to clinics but not in too much detail also add treatment management and symptoms that confirm thalassemia
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Searching PubMed
thalassemia management treatment guidelines
Loading Image
Reading File
I now have comprehensive content from authoritative sources. Let me compose the full answer.
Thalassemia: From Basic to Clinical
1. What Is Thalassemia?
Thalassemia is a group of inherited hemoglobin disorders caused by germline mutations that reduce or abolish the synthesis of either alpha (α) or beta (β) globin chains. The word comes from Greek - thalassa means "sea," reflecting its high prevalence around the Mediterranean basin.
Normal adult hemoglobin (HbA) is a tetramer made of 2 α-chains + 2 β-chains. When one chain is deficient, two problems occur simultaneously:
- Not enough hemoglobin is made (hypochromic, microcytic red cells)
- The other chain builds up in excess, precipitates, and damages red cell membranes - causing hemolysis
- Robbins, Cotran & Kumar Pathologic Basis of Disease
2. Genetics - Where the Mutations Are
| Chain | Gene Location | Mutation Type |
|---|---|---|
| α-globin | Chromosome 16 (2 genes per chromosome = 4 total) | Mainly gene deletions |
| β-globin | Chromosome 11 (1 gene per chromosome = 2 total) | Mainly point mutations (>100 types) |
β-Thalassemia Mutations (3 classes):
- Splicing mutations - most common cause of β+ (reduced output)
- Promoter region mutations - reduce transcription by 75-80%
- Chain terminator mutations - most common cause of β0 (zero output); nonsense or frameshift mutations
Two key mutation categories:
-
β0 = no β-globin produced at all
-
β+ = reduced but detectable β-globin produced
-
Robbins & Kumar Basic Pathology
3. Pathophysiology
The chain imbalance is the root of all the pathology:
Excess unpaired α-chains
↓
Precipitate inside RBC precursors
↓
Membrane damage → Intravascular + extravascular hemolysis
↓
Massive ineffective erythropoiesis in bone marrow
↓
Bone marrow expansion → skeletal deformities
↓
Compensatory extramedullary hematopoiesis → hepatosplenomegaly
↓
Increased GI iron absorption + transfusion iron → Iron overload
In α-thalassemia, excess β-chains form HbH (β4 tetramers), which has extremely high oxygen affinity and delivers little O2 to tissues. In the fetus, excess γ-chains form Hemoglobin Bart (γ4) with the same problem.
- Robbins, Cotran & Kumar; Goldman-Cecil Medicine
4. Classification & Syndromes
β-Thalassemia Spectrum
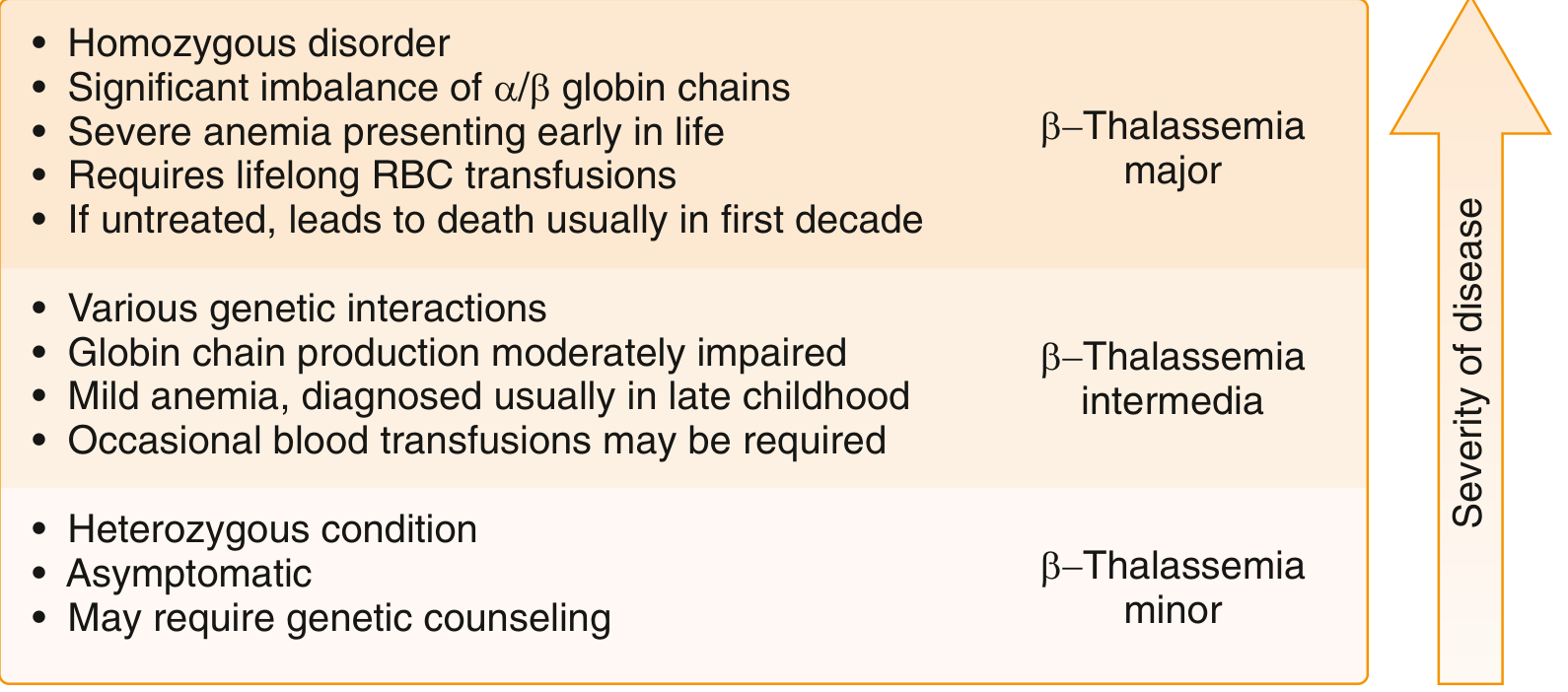
| Form | Genotype | Hb Level | Key Feature |
|---|---|---|---|
| β-Thalassemia Minor (trait) | Heterozygous (1 abnormal allele) | Mildly low or normal | Asymptomatic carrier |
| β-Thalassemia Intermedia | Variable compound heterozygous | 7-10 g/dL | Moderate anemia, no regular transfusions needed |
| β-Thalassemia Major (Cooley's anemia) | Homozygous (both alleles abnormal) | < 7 g/dL | Severe, transfusion-dependent, presents < 2 years |
α-Thalassemia Spectrum (4 gene deletions possible)
| Genes Deleted | Syndrome | Clinical Picture |
|---|---|---|
| 1 (-/αα) | Silent carrier | Completely asymptomatic, slight microcytosis |
| 2 (--/αα or -α/-α) | α-Thalassemia trait | Resembles β-thal minor - microcytosis, minimal anemia |
| 3 (--/-α) | HbH disease | Moderate-severe hemolytic anemia, Hb ~7-9 g/dL |
| 4 (--/--) | Hydrops fetalis (α-thal major) | Fatal in utero without intervention |
- Robbins, Cotran & Kumar
5. Clinical Symptoms That Confirm Thalassemia
Thalassemia Minor/Trait
- Usually asymptomatic - discovered incidentally on CBC
- Mild microcytosis (low MCV) with normal or near-normal hemoglobin
- HbA2 > 3.5% on HPLC is the hallmark of β-thalassemia trait
Thalassemia Intermedia
- Chronic fatigue, pallor, mild jaundice
- Moderate splenomegaly
- Presents in late childhood (after age 2)
- Growth can be mildly retarded
Thalassemia Major (Cooley's Anemia) - Classic Presentation
Children present in the first 1-2 years of life with:
Anemia-related:
- Severe pallor, profound fatigue, poor feeding
- Growth retardation and failure to thrive
- Hemoglobin < 7 g/dL
Expansion of erythroid marrow (bony changes):
- "Crew cut" skull on X-ray (from marrow expansion)
- "Chipmunk facies" - maxillary overgrowth, prominent forehead, frontal bossing
- Pathological fractures
Hepatosplenomegaly:
- Massive splenomegaly (from extramedullary hematopoiesis and red cell trapping)
- Abdominal distension
Jaundice - from chronic hemolysis
Iron overload signs (in transfused/older patients):
- Skin bronzing (hemosiderin deposition)
- Cardiac failure (most common cause of death in poorly managed patients)
- Endocrine: hypogonadism, delayed puberty, diabetes mellitus, hypothyroidism
- Liver: progressive fibrosis and cirrhosis
HbH disease:
-
Jaundice, splenomegaly, hypochromic microcytic anemia
-
"Golf ball" inclusions in RBCs with brilliant cresyl blue stain
-
Goldman-Cecil Medicine; Robbins Pathology
6. Diagnosis
| Test | Finding |
|---|---|
| CBC | Low MCV (< 70 fL), low MCH, microcytic hypochromic anemia |
| Peripheral smear | Target cells, tear-drop cells, basophilic stippling, nucleated RBCs |
| Hemoglobin electrophoresis / HPLC | ↑ HbA2 (> 3.5%) in β-thal trait; ↑ HbF; absent/reduced HbA in major |
| Serum ferritin + iron studies | Normal/elevated (distinguishes from iron deficiency) |
| Genetic testing | Confirms specific mutations |
| Prenatal | Chorionic villus sampling or amniocentesis in at-risk couples |
7. Treatment & Management
Thalassemia Minor
- No specific treatment needed
- Genetic counseling (important to identify at-risk couples)
- Folic acid supplementation during pregnancy
- Iron only if true co-existing iron deficiency is confirmed
HbH Disease
- Folic acid 2-5 mg/day (especially in children)
- Blood transfusions during hemolytic crises (infections can precipitate them)
- Avoid oxidant drugs (e.g., primaquine, dapsone)
Thalassemia Major - Comprehensive Management
1. Regular Transfusion Therapy
- Every 2-5 weeks to maintain pre-transfusion Hb > 9-10.5 g/dL
- Use leukoreduced packed red cells to minimize reactions and pathogen transmission
- Maintains growth, prevents bone deformities
2. Iron Chelation Therapy (mandatory with transfusions)
- Deferoxamine (DFO) - subcutaneous infusion, 8-12 hrs/day, 5-7 days/week
- Deferasirox - oral, once daily; preferred for convenience
- Deferiprone - oral; has added benefit of cardiac iron removal
- Monitoring: serum ferritin, MRI liver and cardiac iron
3. Splenectomy
- Considered when hypersplenism causes excessive transfusion needs
- Optimal management can delay or avoid the need
- Post-splenectomy: penicillin prophylaxis + vaccinations (pneumococcal, meningococcal, Hib)
4. Hematopoietic Stem Cell Transplantation (HSCT)
- The only proven curative option
- Best outcomes in young, well-chelated patients with matched sibling donor
- Also curative for α-thalassemia major (hydrops fetalis survivors on intrauterine transfusion)
5. Folic Acid
- Supplementation for all forms with significant hemolysis
6. Emerging Therapies
-
Gene therapy - shows promise; luspatercept (targets ineffective erythropoiesis) is FDA-approved for transfusion-dependent β-thalassemia in adults
-
Regular endocrine, cardiac, and hepatic monitoring and treatment of iron overload complications
-
Goldman-Cecil Medicine, International Edition; Creasy & Resnik's Maternal-Fetal Medicine
8. Epidemiology & Why It Persists
Thalassemia is most common in the Mediterranean basin, Middle East, tropical Africa, Indian subcontinent, and Southeast Asia - precisely the malaria belt. Heterozygous carriers have a selective survival advantage against P. falciparum malaria, which explains why these mutations have remained so common in the population despite their harmful effects in homozygotes. - Robbins, Cotran & Kumar
Quick Summary Table
| Feature | Minor | Intermedia | Major |
|---|---|---|---|
| Anemia | Mild/none | Moderate (7-10 g/dL) | Severe (< 7 g/dL) |
| Transfusions | Not needed | Occasional | Lifelong (every 2-5 weeks) |
| Iron chelation | Rarely | Sometimes | Always |
| Bony changes | No | Mild | Severe (chipmunk facies, crew-cut skull) |
| Hepatosplenomegaly | No | Moderate | Severe |
| Curative option | N/A | HSCT if severe | HSCT |
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease; Robbins & Kumar Basic Pathology; Goldman-Cecil Medicine International Edition; Creasy & Resnik's Maternal-Fetal Medicine.
This is a shared conversation. Sign in to Orris to start your own chat.