Pathophysiology of high blood pressure
Reading File
Finding Sources
Finding Sources
Searching PubMed
"hypertension"[MeSH Terms] AND "pathophysiology"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive textbook content. Let me compose the full answer.
Pathophysiology of High Blood Pressure (Hypertension)
Hypertension is defined as a sustained elevation in blood pressure (BP) above 130/80 mmHg (ACC/AHA 2017 guidelines) or 140/90 mmHg (older/WHO definition). Over 90% of cases are primary (essential) hypertension, where no single reversible cause is identified. The remaining ~10% are secondary hypertension (renal artery stenosis, primary aldosteronism, pheochromocytoma, etc.).
1. The Fundamental Formula
BP = Cardiac Output (CO) × Total Peripheral Resistance (TPR)
Long-term BP regulation requires more than just this formula. It also depends on:
- Kidney regulation of body fluid and sodium balance
- Local autoregulation of tissue blood flow
- Time-dependent interactions between multiple control systems
In most patients with established primary hypertension, CO is normal and TPR is elevated - not as a primary driver, but as an autoregulatory response to maintain normal tissue perfusion at higher pressures.
- Fuster and Hurst's The Heart, 15th Ed.
2. Renal Mechanisms and Pressure Natriuresis
The kidney is the most important long-term regulator of BP. It does this via pressure natriuresis: a rise in mean arterial pressure (MAP) of as little as 1-3 mmHg causes the kidney to increase sodium and water excretion, reducing blood volume and returning BP toward normal.
How it works:
- Rising renal perfusion pressure increases medullary blood flow (less tightly autoregulated than cortical flow), which washes out medullary interstitial NaCl, reducing the osmotic gradient for tubular water reabsorption
- Elevated pressure reduces proximal tubular reabsorption by altering peritubular capillary Starling forces (lower oncotic pressure, higher hydrostatic pressure)
How it breaks down in hypertension:
-
In essential hypertension, the pressure-natriuresis curve is shifted rightward - the kidney requires a higher BP set-point to excrete a given sodium load
-
This shift can result from: increased renal sympathetic activity, excess angiotensin II, structural nephron loss, reduced nitric oxide generation, or oxidative stress
-
Salt sensitivity (BP rises with sodium intake) is present in ~30% of normotensives and ~60% of hypertensives, and is especially prevalent in Black individuals, the elderly, and those with chronic kidney disease (CKD)
-
National Kidney Foundation Primer on Kidney Diseases, 8e
3. The Renin-Angiotensin-Aldosterone System (RAAS)
The RAAS is a central pressor pathway:
- Decreased renal perfusion or sodium delivery → juxtaglomerular cells secrete renin
- Renin cleaves angiotensinogen → Angiotensin I
- ACE converts Ang I → Angiotensin II (Ang II)
- Ang II acts on:
- Vascular smooth muscle (AT1 receptors): potent vasoconstriction → raises TPR
- Adrenal cortex: stimulates aldosterone secretion → sodium and water retention → raises CO
- Renal tubules: directly promotes sodium reabsorption
- SNS: facilitates norepinephrine release, amplifying sympathetic tone
- CNS: stimulates thirst and ADH release
In primary hypertension, renin levels are variable (low, normal, or high), meaning RAAS overactivation is not universal, but it remains a key amplifier of hypertension, especially when volume expansion is already present.
4. Sympathetic Nervous System (SNS) Activation
Increased sympathetic activity contributes to hypertension via:
- Cardiac: increased heart rate and contractility → raises CO
- Vascular: alpha-1 receptor-mediated vasoconstriction → raises TPR
- Renal: enhanced tubular NaCl reabsorption + increased renin release from juxtaglomerular apparatus → sodium retention
Obesity is a key driver of SNS activation: adipose tissue increases leptin, which directly stimulates central sympathetic outflow. This explains why overweight and obesity account for 65-75% of the risk for primary hypertension.
- Fuster and Hurst's The Heart, 15th Ed.
5. Vascular Mechanisms
Endothelial Dysfunction
- Normal endothelium produces nitric oxide (NO) via nitric oxide synthase (NOS), which causes vasodilation and inhibits tubular NaCl reabsorption
- In hypertension, NOS activity is reduced, causing impaired vasodilation and increased peripheral resistance
- Reactive oxygen species (ROS) - especially superoxide (O₂⁻) - inactivate NO by forming peroxynitrite (ONOO⁻), further reducing vasodilatory capacity and promoting oxidative vascular injury
Vascular Remodeling
- Chronic elevated BP triggers hypertrophic remodeling of vessel walls: smooth muscle hypertrophy, increased wall-to-lumen ratio
- Capillary rarefaction (reduced capillary density) occurs in tissues, raising TPR structurally
- The autoregulatory curve shifts rightward in chronic hypertension, meaning lower BPs are now required to maintain adequate perfusion - rapid BP lowering can cause ischemia
Endothelin
-
Produced by vascular endothelium and collecting tubules
-
ET-A receptors mediate vasoconstriction; ET-B receptors promote NO release and natriuresis
-
ET-A overactivation contributes to increased SVR, especially in volume-expanded and CKD-related hypertension
-
National Kidney Foundation Primer on Kidney Diseases, 8e; Fuster and Hurst's The Heart, 15th Ed.
6. Blood Flow Autoregulation in Hypertension
The body regulates tissue blood flow acutely (seconds-minutes) via:
- Myogenic response: vessels constrict when stretched by high pressure
- Metabolic response: vasodilation in response to local hypoxia/CO₂ accumulation
In hypertension, this autoregulatory range shifts to higher pressures (see figure below). This protects organs from overperfusion short-term but means BP must be lowered gradually with antihypertensive therapy to avoid ischemia.
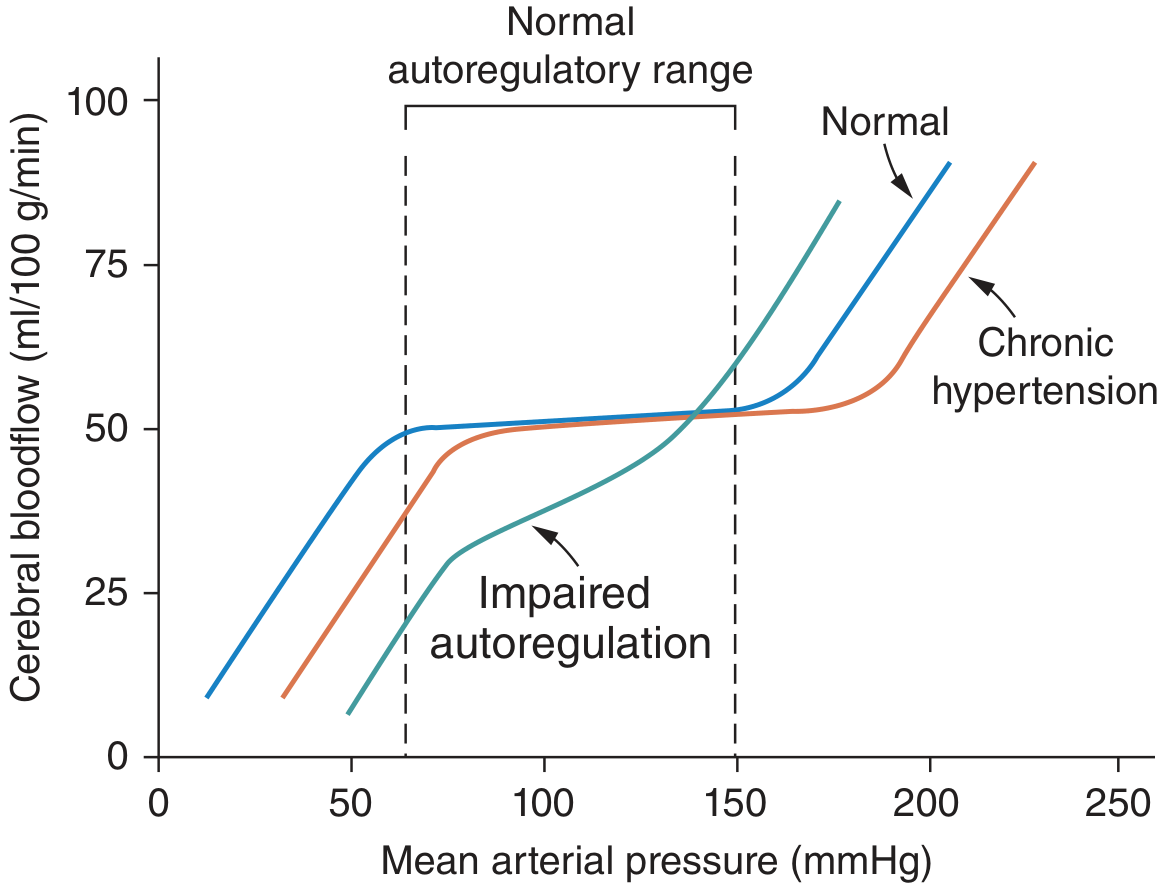
7. Hormonal & Humoral Modulators
| Factor | Effect on BP | Mechanism |
|---|---|---|
| Aldosterone | Raises | Sodium retention via ENaC in collecting duct |
| ANP (Atrial Natriuretic Peptide) | Lowers | Increases GFR, inhibits renin, promotes natriuresis |
| Endothelin-1 | Raises (ET-A) | Vasoconstriction, sodium retention |
| Nitric Oxide | Lowers | Vasodilation, natriuresis |
| Prostaglandins (PGI₂) | Lowers | Vasodilation |
| Thromboxane A₂ | Raises | Vasoconstriction, platelet aggregation |
| Reactive Oxygen Species | Raises | Inactivate NO, promote vasoconstriction |
| NSAIDs (COX inhibition) | Raises | Block vasodilatory prostaglandins → sodium retention |
- National Kidney Foundation Primer on Kidney Diseases, 8e
8. Obesity-Hypertension Nexus
Obesity causes hypertension through multiple parallel pathways:
- Renal compression by perirenal fat → impaired pressure natriuresis
- SNS activation via leptin hypersecretion
- RAAS activation (adipose tissue expresses local renin-angiotensin components)
- Insulin resistance/hyperinsulinemia → promotes tubular sodium reabsorption and SNS activity
- Obstructive sleep apnea (common in obesity) → intermittent hypoxia → SNS surges → sustained daytime hypertension
9. Genetic Causes (Secondary/Monogenic)
Rare but well-characterized Mendelian causes illustrate the importance of sodium handling:
| Disorder | Mechanism | Key Features |
|---|---|---|
| Liddle Syndrome | Gain-of-function ENaC mutation | Low aldosterone, low K⁺, responds to amiloride |
| Gordon Syndrome | Excess NaCl reabsorption (WNK kinase mutations) | High K⁺, responds to thiazides |
| Glucocorticoid-remediable aldosteronism (FH-I) | Aldosterone under ACTH control | High aldosterone, responds to dexamethasone |
| AME (Apparent Mineralocorticoid Excess) | 11β-HSD2 deficiency; cortisol acts on MR | Low aldosterone, low K⁺ |
- Fuster and Hurst's The Heart, 15th Ed.
10. Hypertension in CKD (Vicious Cycle)
CKD creates a self-reinforcing cycle:
- Nephron loss → reduced natriuretic capacity → salt sensitivity and volume expansion
- Volume expansion → higher BP → further glomerular hypertension and injury
- Injured glomeruli → additional nephron loss → worsening hypertension
- CKD also: increases endothelin, reduces NO (via ADMA accumulation), amplifies SNS discharge via abnormal afferent renal nerve signals, and promotes oxidative stress
Summary: Integrated Pathophysiology
Genetic predisposition + environmental factors (obesity, excess salt, sedentary lifestyle)
↓
Impaired pressure-natriuresis (kidney set-point shifted rightward)
↓
Sodium and water retention → ↑ blood volume → ↑ CO
↓
↑ Ang II, SNS activation, endothelin
↓
Vasoconstriction + vascular remodeling → ↑ TPR
↓
Endothelial dysfunction (↓ NO, ↑ ROS) → further ↑ TPR
↓
SUSTAINED HYPERTENSION
↓
Target-organ damage: LVH, nephrosclerosis, atherosclerosis, stroke
The kidney sits at the center of long-term BP control - ultimately, sustained hypertension requires a defect in renal sodium excretion, whether primary (essential) or secondary (CKD, RAAS excess, structural renal disease).
This is a shared conversation. Sign in to Orris to start your own chat.