Acute tubular necrosis of kidney
Reading File
Finding Sources
Searching PubMed
"acute tubular necrosis" AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have all the material needed for a thorough, well-illustrated answer. Here it is:
Acute Tubular Necrosis (ATN) / Acute Tubular Injury (ATI)
Terminology note: Pathologists now prefer Acute Tubular Injury (ATI) over ATN because frank necrosis is often absent on biopsy; necrosis can be focal or entirely missed in a cortical sample. Clinically and in most texts the term ATN persists.
1. Overview and Epidemiology
ATN is the most common cause of acute kidney injury (AKI) in hospitalized patients, accounting for ~50% of all in-hospital AKI cases and >50% of ICU-acquired AKI. It is characterized by an abrupt, sustained decline in GFR triggered by an ischemic or nephrotoxic insult, developing within minutes to days after the event.
- Robbins Pathologic Basis of Disease, p. 863
- Frameworks for Internal Medicine, p. 454
2. Causes
| Type | Mechanisms / Examples |
|---|---|
| Ischemic ATN | Shock (septic, cardiogenic, hypovolemic), severe trauma, acute pancreatitis, burns, major surgery, prolonged hypotension |
| Nephrotoxic ATN | Aminoglycosides (gentamicin), radiocontrast agents, amphotericin B, cisplatin, heavy metals (mercury, lead), organic solvents (carbon tetrachloride) |
| Pigment nephropathy | Myoglobinuria (rhabdomyolysis/crush injury), hemoglobinuria (mismatched transfusion, hemolytic crises) |
| Protein injury | Multiple myeloma (Bence-Jones proteins/light chains) |
| Transplant-related | Prolonged cold ischemia time, ischemia-reperfusion injury in allografts causing delayed graft function (DGF) |
- Robbins, p. 863-864
- Frameworks for Internal Medicine, p. 455
3. Pathophysiology
Two parallel events are central to ATN: tubular epithelial cell injury and persistent hemodynamic disturbances.
3a. Tubular Cell Injury
The S3 segment of the proximal tubule (straight portion, in the outer medullary stripe) is the most vulnerable cell because:
- It depends almost exclusively on fatty acid oxidation (minimal capacity for anaerobic glycolysis)
- It receives primarily venous capillary flow, so medullary perfusion fails to recover even when cortical flow normalizes ("extension phase")
Sequence of injury events:
- Loss of brush border - earliest change; microvilli disruption with bleb formation into the lumen
- Loss of cell polarity - Na⁺/K⁺-ATPase redistributes from the basolateral to the apical/luminal surface → defective Na⁺ reabsorption → increased distal Na⁺ delivery
- Tubuloglomerular feedback (TGF) - elevated distal Na⁺ triggers afferent arteriolar vasoconstriction, dropping GFR further
- Cell detachment - necrotic/apoptotic cells slough from the basement membrane, form casts, and obstruct the tubular lumen → increased intratubular pressure → further GFR reduction
- Tubular backleak - areas of denuded basement membrane allow filtered urine to leak back into the interstitium → interstitial edema → increased interstitial pressure → worsens tubular obstruction
- Inflammation - injured cells release cytokines and DAMPs, recruit neutrophils, macrophages, NKT cells, and T lymphocytes → amplify injury
In toxic ATN, S1/S2 segments of the proximal convoluted tubule are additionally targeted because of their high rates of endocytosis and active transport, leading to concentrated intracellular accumulation of the toxin.
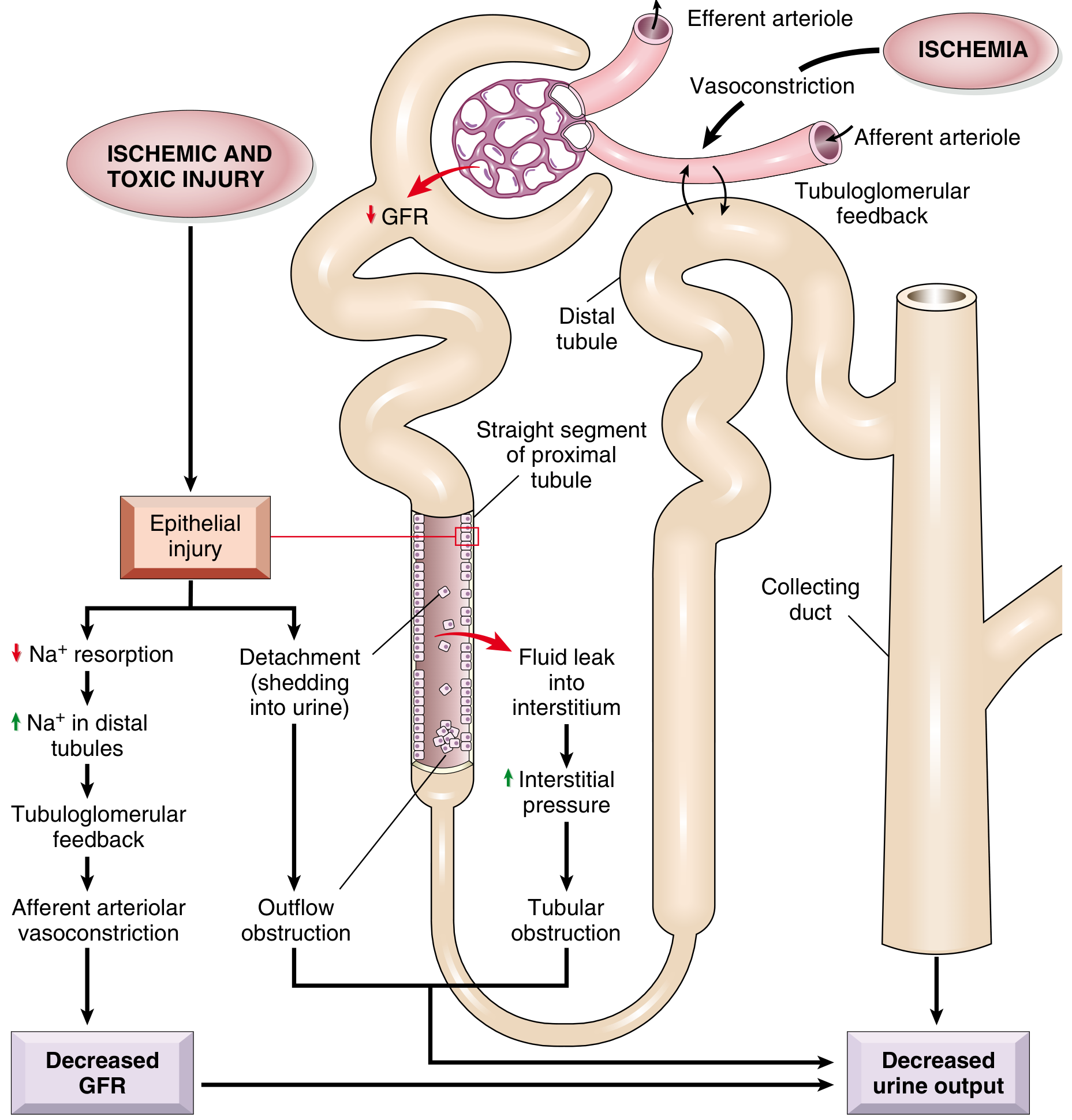
3b. Vascular/Hemodynamic Disturbances
- Decreased synthesis and/or increased consumption of vasodilatory prostaglandins and nitric oxide in the ischemic kidney
- Increased endothelin and angiotensin II-mediated vasoconstriction
- Endothelial swelling, leukocyte adhesion, and platelet aggregation in peritubular capillaries perpetuate medullary ischemia even after the initial insult resolves
ATN has been described as a "(mal)adaptive response" - the kidney "trades away" GFR to preserve medullary oxygenation and tubular integrity.
- Comprehensive Clinical Nephrology 7e, p. 990
- Brenner and Rector's The Kidney, p. 1205
4. Morphology / Histology
Pattern of injury (Ischemic vs. Toxic)
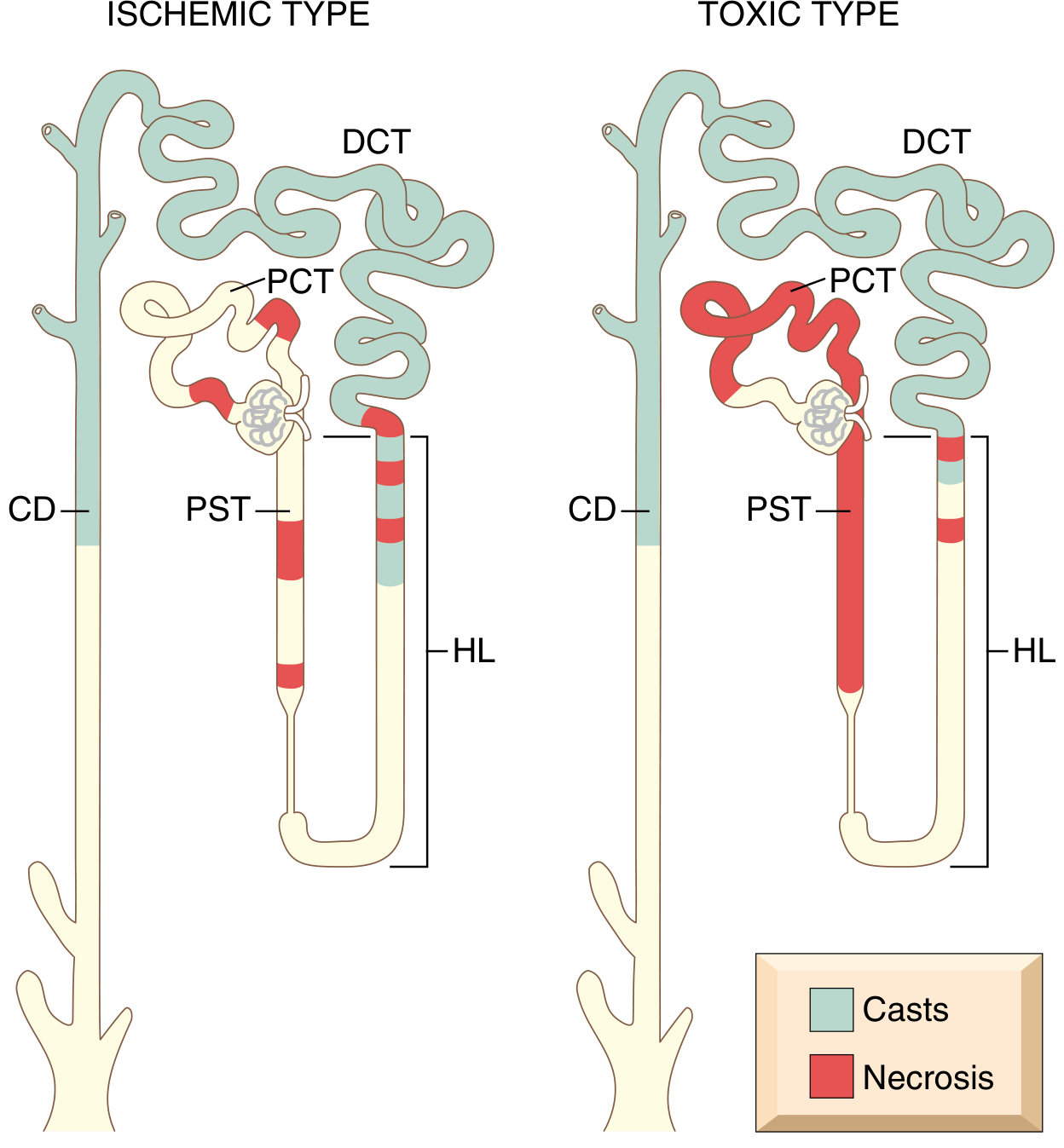
| Feature | Ischemic ATN | Toxic ATN |
|---|---|---|
| Distribution | Patchy, skip areas, short lengths | Extensive, more uniform |
| Primary site | PST (S3), outer medullary thick ascending limb | PCT (S1, S2) |
| Basement membrane | Often ruptured (tubulorrhexis) | May remain intact |
| Casts | DCT and collecting duct | DCT and collecting duct |
| Special features | Interstitial edema, leukocytes in vasa recta | Specific changes with toxin (e.g., acidophilic inclusions in mercuric chloride; oxalate crystals in ethylene glycol) |
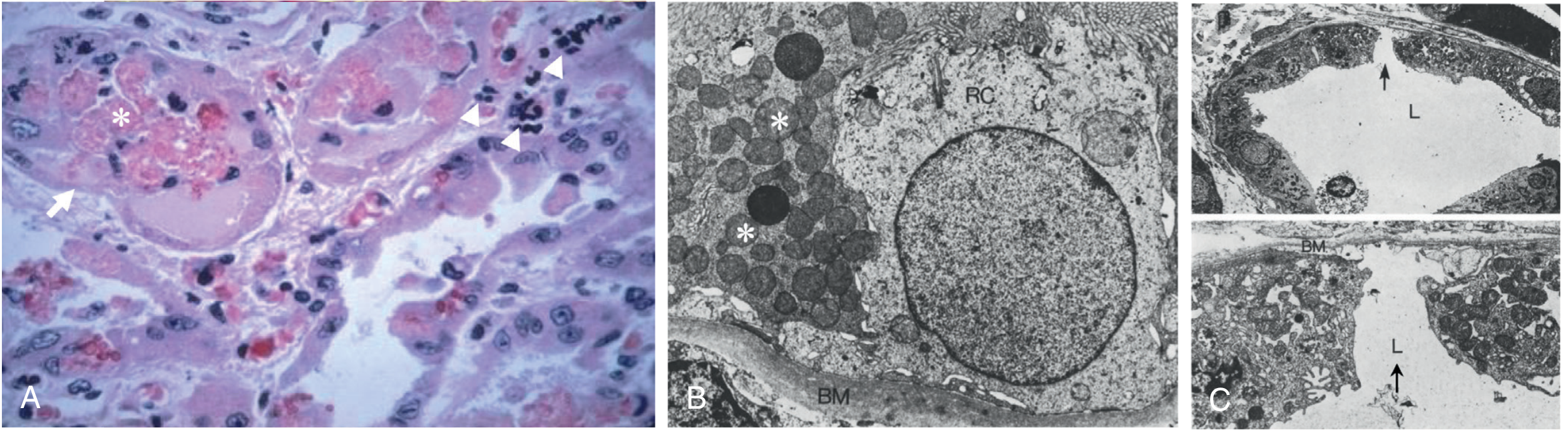
Key histologic findings (human biopsy):
- Vacuolation and loss of brush border in proximal tubule cells
- Thinning of epithelium to maintain tubular monolayer integrity
- Intraluminal cast accumulation with cellular debris
- Apoptosis of tubular cells (necrosis may be subtle or focal)
- Regenerative changes: flattened cells, hyperchromatic nuclei, mitotic figures - coexisting with fresh injury in the same biopsy (indicating ongoing ischemic episodes during the maintenance phase)
- Necrosis is focal and often missed on biopsy because the outer medulla is inadequately sampled
- Robbins, p. 864-865
- Comprehensive Nephrology 7e, p. 990-991
5. Clinical Features - The Three Phases
| Phase | Duration | Key Features |
|---|---|---|
| Initiation | ~36 hours | Dominated by precipitating event; modest oliguria; slight rise in BUN - easily mistaken for prerenal AKI |
| Maintenance (oliguric) | Days to weeks | Urine output 40-400 mL/day; rising BUN and creatinine; hyperkalemia; metabolic acidosis; fluid/electrolyte overload; uremia; risk of death highest here |
| Recovery (polyuric) | Gradual | Urine output may reach 3 L/day as tubular function lags behind glomerular recovery; risk of hypokalemia and hyponatremia; BUN and creatinine gradually normalize |
- ~50% of ATN is non-oliguric (urine output >400 mL/day), especially nephrotoxic ATN; this carries a better prognosis.
- Robbins, p. 864-865
6. Diagnosis
Urinalysis and Urine Microscopy
The most characteristic finding is "muddy brown" coarse granular casts - ATN is highly likely when ≥6 granular casts are seen per high-power field.

Laboratory Indices
| Test | ATN | Prerenal AKI |
|---|---|---|
| FENa | >1-2% | <1% |
| FEUrea (on diuretics) | >35-50% | <35% |
| Urine Na | >40 mEq/L | <20 mEq/L |
| Urine osmolality | ~300 mOsm/kg (isosthenuria) | >500 mOsm/kg |
| Urine specific gravity | ~1.010 | >1.020 |
| BUN:Creatinine ratio | ~10-15:1 | >20:1 |
| Urine sediment | Muddy brown granular casts, tubular epithelial cells | Hyaline casts, bland |
- Comprehensive Nephrology 7e, p. 990
- Frameworks for Internal Medicine, p. 454-455
Note: FENa can be falsely low (<1%) in ATN superimposed on: contrast nephropathy, myoglobinuria, early obstruction, or hepatorenal syndrome. FEUrea is more reliable in patients on diuretics.
Novel Biomarkers
- Urine NGAL (neutrophil gelatinase-associated lipocalin), KIM-1 (kidney injury molecule-1), IL-18, TIMP-2 x IGFBP7 (NephroCheck) - can detect ATN before creatinine rises
- No definitive non-invasive test exists; kidney biopsy is the gold standard but rarely needed
7. Management
Management is largely supportive - no pharmacologic agent has proven effective in altering the course of established ATN in humans.
Supportive Care
- Treat the underlying cause - optimize hemodynamics, discontinue nephrotoxins, treat sepsis
- Fluid balance - avoid fluid overload; restrict intake to insensible losses + urine output
- Electrolytes - manage hyperkalemia (ECG monitoring, calcium gluconate, insulin/glucose, kayexalate, dialysis if refractory); correct metabolic acidosis
- Nutrition - adequate protein/calorie intake (avoid excessive protein restriction)
- Renal replacement therapy (RRT) - indicated for refractory hyperkalemia, acidosis, fluid overload, uremic symptoms; continuous RRT preferred in hemodynamically unstable patients
- Drug dose adjustment - renally clear drugs must be dosed per reduced GFR
Pharmacologic Interventions - What Does NOT Work
The following agents have been tested in clinical trials and found ineffective for established ATN:
- Loop diuretics (furosemide) - no change in course; may convert oliguric to non-oliguric ATN but does not improve outcomes
- Low-dose ("renal dose") dopamine - does not prevent or shorten ATN (increases renal vascular resistance in patients with AKI)
- Fenoldopam, atrial natriuretic peptide, thyroid hormone, alpha-MSH, erythropoietin, insulin growth factor, prostaglandin A1
Prevention
-
Pre-procedure volume expansion (IV saline before contrast administration or nephrotoxic drugs)
-
N-acetylcysteine - evidence mixed, but low risk; commonly used before radiocontrast
-
Minimize aminoglycoside exposure; use once-daily dosing with therapeutic drug monitoring
-
Remote ischemic preconditioning (RIPC) - brief ischemia to a limb triggers HIF-1α, heat shock proteins, and protective signaling; promising but not yet standard of care
-
Brenner and Rector's The Kidney, p. 1268-1269
8. Prognosis
- ATN is generally reversible if the precipitating cause is corrected - recovery depends on intact tubular basement membranes allowing re-epithelialization
- Hospital mortality for ICU-associated ATN requiring dialysis: 50-80%
- Incomplete recovery occurs in ~30% of survivors, leading to CKD or end-stage renal disease
- Patchiness of necrosis and preserved basement membrane segments are the structural basis for recovery
Summary Diagram - Pathophysiology
The four key mechanisms converging on reduced GFR and oliguria in ATN:
- Afferent vasoconstriction (via TGF from defective Na⁺ reabsorption)
- Tubular cast obstruction (from sloughed epithelial cells)
- Tubular backleak (filtrate re-enters interstitium through denuded basement membrane)
- Interstitial edema and pressure (from backleak, compressing tubules and capillaries)
Sources:
- Robbins, Cotran & Kumar - Pathologic Basis of Disease, p. 863-865
- Brenner and Rector's The Kidney (2-Volume Set), p. 1205-1210, 1268-1269
- Comprehensive Clinical Nephrology, 7th Edition, p. 989-991
- Frameworks for Internal Medicine, p. 454-455
Acute tubulointerstitial nephritis
Finding Sources
Searching PubMed
"tubulointerstitial nephritis" AND "acute interstitial nephritis"
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Excellent - I have all the material I need including the biopsy images. Now let me compile the comprehensive answer.
Acute Tubulointerstitial Nephritis (ATIN / AIN)
1. Definition
Acute tubulointerstitial nephritis (ATIN) is an inflammatory disorder of the renal tubules and interstitium characterized histologically by interstitial inflammatory infiltrate, tubulitis, variable tubular injury, and interstitial edema, with an acute - often reversible - decline in GFR developing over days to weeks. When recognized and treated late, it can transition to chronic interstitial nephritis with interstitial fibrosis and tubular atrophy.
- Brenner and Rector's The Kidney, p. 1595
- Harrison's Internal Medicine 22E, p. 1457
2. Epidemiology
-
Accounts for 1-3% of all kidney biopsies and 10-20% of biopsies for unexplained AKI
-
Represents 15-27% of biopsies performed for AKI in some series
-
Its incidence appears to be rising, in part due to increasing polypharmacy and immune checkpoint inhibitor use
-
More common in older individuals (drug-induced) vs. younger individuals (immune-mediated, TINU)
-
Medications are now the leading cause in developed countries
-
Brenner and Rector's The Kidney, p. 1595-1596
-
Comprehensive Clinical Nephrology 7e, p. 889
3. Etiology and Classification
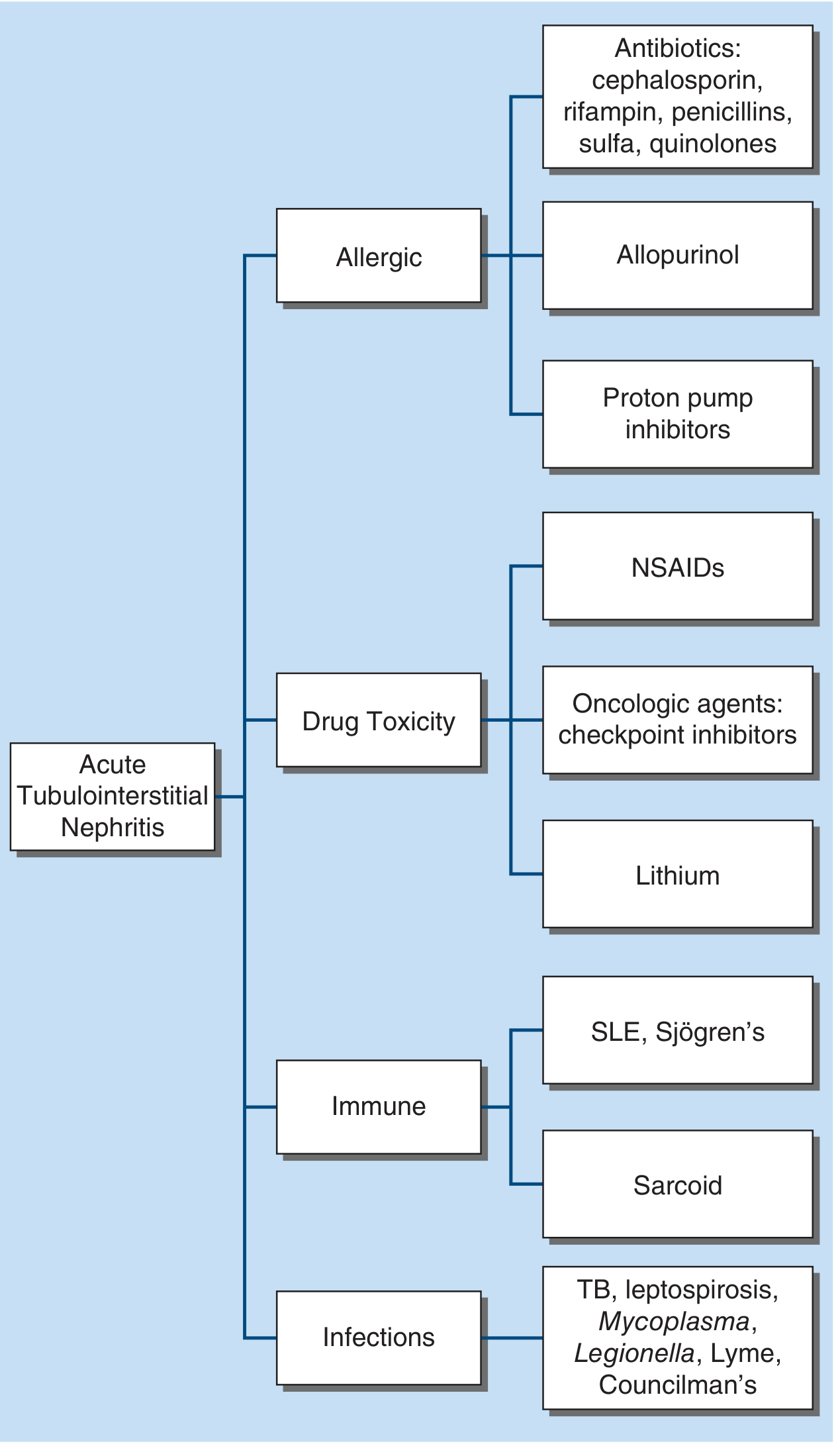
A. Drug-Induced (Most Common - ~70% of all ATIN)
In a large biopsy series: antibiotics 49%, PPIs 14%, NSAIDs 11%; top individual drugs: omeprazole (12%), amoxicillin (8%), ciprofloxacin (8%).
| Drug Class | Examples | Key Features |
|---|---|---|
| β-Lactam antibiotics | Penicillins (methicillin, amoxicillin), cephalosporins | Classic presentation; fever/rash/eosinophilia in >75%; short latency (days to weeks) |
| Sulfonamides | Sulfamethoxazole-trimethoprim | Hypersensitivity syndrome; higher rate in HIV, transplant patients |
| Fluoroquinolones | Ciprofloxacin, levofloxacin | Rarely associated with hypersensitivity syndrome |
| Rifampin | - | Dose-dependent; anti-rifampin antibodies; hemolytic anemia, thrombocytopenia; dialysis required in ~2/3 |
| Proton Pump Inhibitors | Omeprazole, lansoprazole, pantoprazole | Long latency (mean 11 weeks to months); few systemic allergic features; all PPI classes implicated |
| NSAIDs | All NSAIDs, especially fenoprofen | Long latency (months); minimal allergic features; may coexist with minimal change disease/heavy proteinuria |
| Antiepileptics | Phenytoin, carbamazepine, valproate | - |
| Diuretics | Thiazides, furosemide, acetazolamide | Sulfa-containing agents |
| Allopurinol | - | Often with DRESS syndrome |
| Immune checkpoint inhibitors | PD-1, PD-L1, CTLA-4 inhibitors | ~3% incidence; average 4 months latency; patients who recover may often resume the agent |
| Antivirals | Acyclovir, indinavir, foscarnet, tenofovir | Rare |
B. Infectious (Pre-antibiotic era was dominant cause)
- Bacterial: Streptococcal infections (Councilman's nephritis in scarlet fever/diphtheria - historical), Legionella, leptospirosis, Mycobacterium tuberculosis (caseating granulomas first appearing near glomeruli in cortex where O₂ tension is highest)
- Viral: EBV, CMV, hantavirus, HIV
- Other: Mycoplasma, Lyme disease, toxoplasmosis
- Mechanism: indirect immune/hypersensitivity reaction; the pathogen is not cultured from the renal parenchyma (distinguishing it from pyelonephritis)
C. Immune-Mediated / Autoimmune
| Condition | Key Features |
|---|---|
| SLE | Hematuria, proteinuria, pyuria, WBC casts; SLE serologies + ; hypocomplementemia; lymphocyte-dominant infiltrate |
| Sjögren syndrome | Renal involvement in 15-67% of cases; lymphocyte-dominant infiltrate with tubulitis; distal RTA is the classic tubulopathy |
| Sarcoidosis | Noncaseating granulomas in interstitium; often with hypercalcemia/hypercalciuria; responds to steroids |
| ANCA vasculitis | ANCA + serology; pauci-immune lesion; mononuclear infiltrate with plasma cells and eosinophils |
| Anti-GBM disease / IgA nephropathy | Concomitant tubulointerstitial lesion common |
| IgG4-related kidney disease | Elevated serum IgG4 (in ~60%); storiform fibrosis on biopsy; multisystem involvement; middle-aged men |
D. Tubulointerstitial Nephritis and Uveitis Syndrome (TINU)
A rare, distinct entity most common in adolescent girls but occurring at any age. Presents with:
-
Uveitis (painful red eyes, photophobia) - may precede, follow, or occur simultaneously with renal disease
-
Systemic features: weight loss, fever, anemia, hyperglobulinemia
-
Isolated proximal tubulopathy or Fanconi syndrome as the initial renal manifestation
-
Associated infections (EBV, toxoplasmosis, giardiasis) have been reported
-
Treatment: steroids for 3-6 months with slow taper; steroid-sparing agents (MMF, methotrexate, cyclosporine) for relapses
-
Brenner and Rector's The Kidney, p. 1604-1605
4. Pathogenesis
ATIN is primarily an immunologically mediated hypersensitivity reaction. Evidence for this includes:
- Occurs in only a small fraction of exposed patients (not dose-dependent for most agents)
- Associated with extrarenal manifestations of hypersensitivity
- Recurs with re-exposure to the same or related drug (anamnestic response)
- Interstitial infiltrates contain T lymphocytes and sometimes granulomas (DTH)
- No neutrophilic (purulent) character to the inflammation
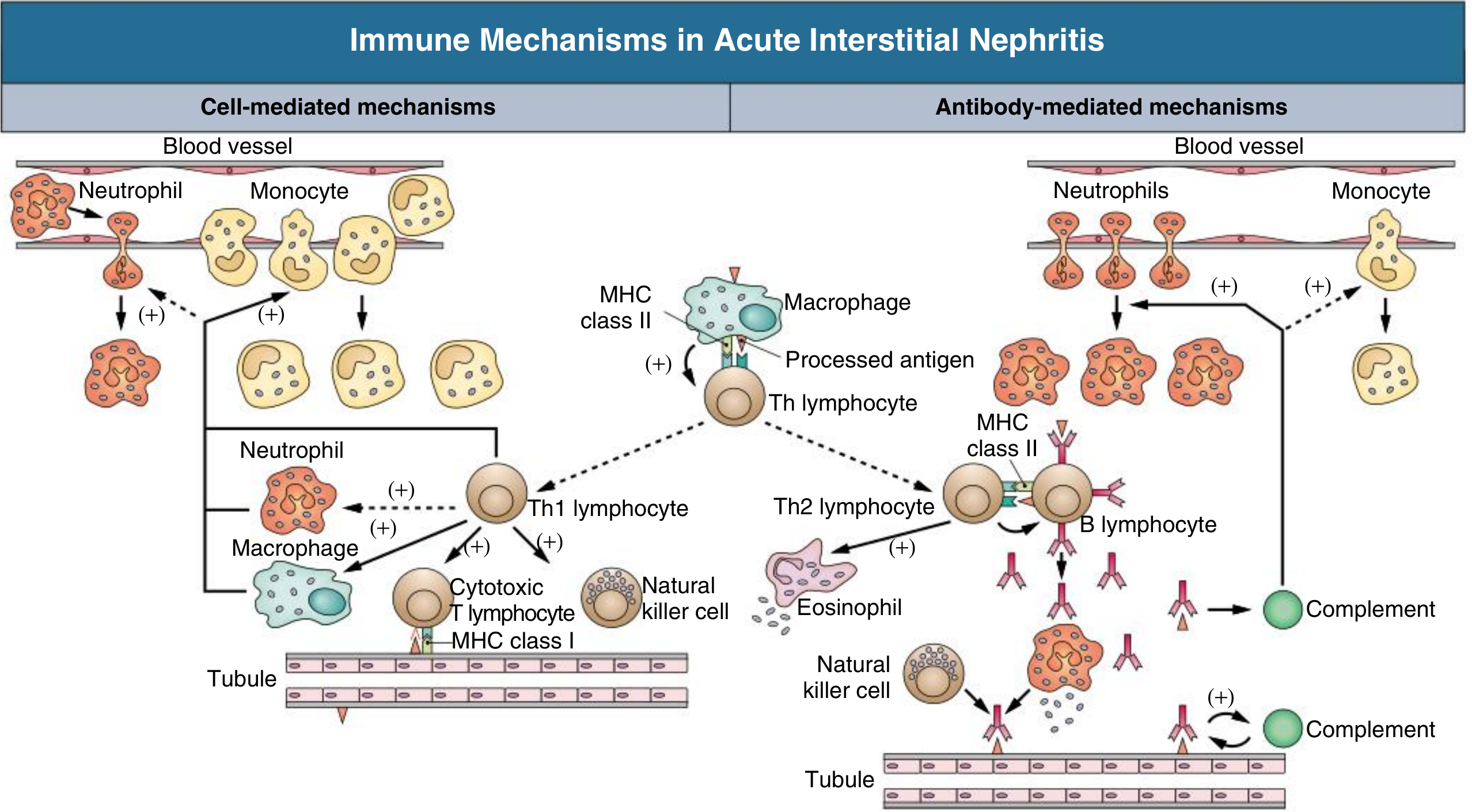
Key Antigens Implicated:
- Tubular basement membrane (TBM) components - glycoproteins 3M-1 and TIN-Ag/TIN1
- Secreted tubular proteins - uromodulin (Tamm-Horsfall protein)
- Non-renal ("planted") antigens - drugs/metabolites binding to kidney structures; acting as haptens modifying native proteins; immune complex deposition within the interstitium
Inflammatory Cascade:
-
Drug/antigen → processed and presented via MHC class II to Th lymphocytes
-
Cell-mediated (dominant): Th1 → cytotoxic CD8+ T cells and macrophages → direct tubular injury
-
Antibody-mediated: Th2 → B cells → anti-TBM antibodies → complement activation + eosinophil recruitment
-
Interstitial infiltration → inflammatory cytokine release (IL-1, TNF, TGF-β) → fibroblast activation → interstitial fibrosis if untreated
-
Comprehensive Clinical Nephrology 7e, p. 889-890
5. Histopathology
Kidney biopsy remains the gold standard for diagnosis.
Key Histologic Features:
- Interstitial inflammatory infiltrate - primarily mononuclear cells; lymphocytes (CD4+ and CD8+ T cells predominant), monocytes/macrophages, plasma cells, B cells
- Tubulitis - lymphocytes crossing the tubular basement membrane and invading the tubular epithelium (pathognomonic)
- Interstitial edema - often prominent, especially early
- Interstitial fibrosis - variable; severity predicts prognosis and likelihood of CKD
- Eosinophils - more common in drug-induced (especially β-lactam) ATIN; often absent in NSAID-induced
- Granulomas - noncaseating in sarcoidosis, drug reactions, and some infections; caseating in TB
- Neutrophils - when prominent, raise concern for pyelonephritis
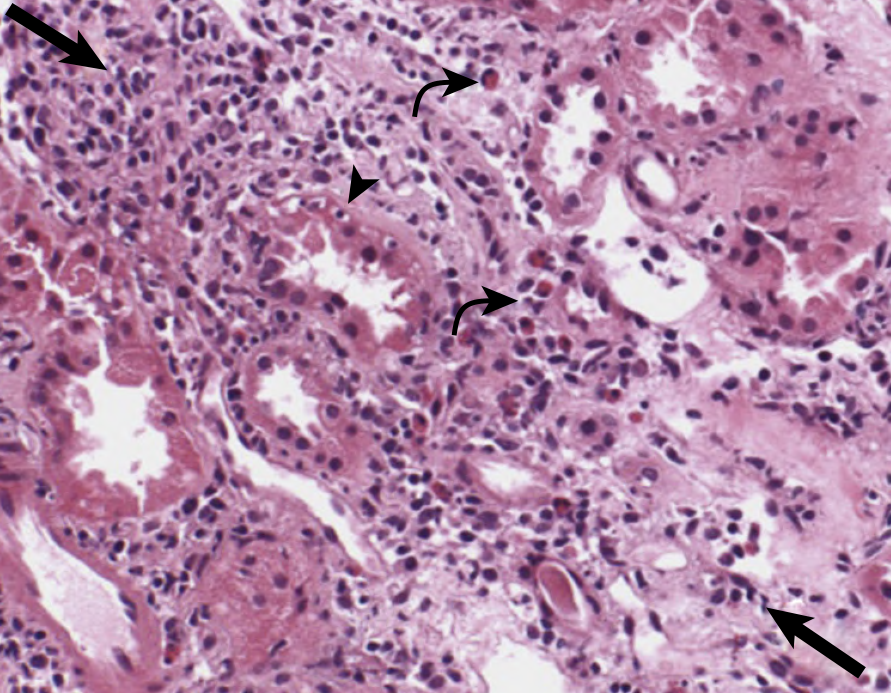
Biopsy Differential by Infiltrate Composition:
| Finding | Suggests |
|---|---|
| Abundant eosinophils | Drug-induced (β-lactams, sulfonamides) |
| Neutrophil predominance | Pyelonephritis |
| Noncaseating granulomas | Sarcoidosis, rifampin, NSAIDs |
| Caseating granulomas | Tuberculosis |
| Minimal-change glomerulopathy co-existing | NSAIDs |
| Plasma cells + storiform fibrosis | IgG4-related disease |
| Lymphocytic tubulitis only | SLE, Sjögren, PPI-induced |
- Brenner and Rector's The Kidney, p. 1599-1600
- NKF Primer on Kidney Diseases 8e
6. Clinical Features
The classically taught triad of fever, maculopapular rash, and peripheral eosinophilia occurs in only 5-15% of patients overall and should not be relied upon to make or exclude the diagnosis. It is more common in antibiotic-induced (especially methicillin/penicillin) ATIN and virtually absent in NSAID-induced ATIN.
Renal Manifestations:
- Acute rise in serum creatinine (often the only finding)
- Oliguria (in severe cases)
- Flank pain (due to acute distension of the renal capsule)
- Polyuria and nocturia (tubular dysfunction - nephrogenic DI)
- Signs of tubular dysfunction: renal tubular acidosis, glycosuria with normal blood glucose, hypophosphatemia, hypokalemia or hyperkalemia, Fanconi syndrome (proximal RTA + glycosuria + phosphaturia + aminoaciduria + bicarbonaturia)
Extrarenal Manifestations (drug-induced):
- Fever (common with β-lactams)
- Maculopapular rash
- Peripheral eosinophilia
- Arthralgias (45% in one analysis)
- DRESS syndrome (Drug Rash, Eosinophilia, and Systemic Symptoms) - associated with AIN in up to 40% of cases with prolonged drug exposure
Temporal Pattern:
-
Drug reaction: days to weeks after starting the drug; can be months later with NSAIDs, PPIs, or checkpoint inhibitors
-
Classic drug reaction setting: febrile patient on antibiotics who defervesces but develops recurrent fever several days later
-
Re-exposure: rapid, severe recurrence (anamnestic response)
-
Comprehensive Clinical Nephrology 7e, p. 890
-
Goldman-Cecil Medicine, p. 3544-3548
-
Frameworks for Internal Medicine, p. 456
7. Diagnosis
Urinalysis and Urine Microscopy
The most diagnostically useful investigation.
| Urinary Finding | Significance |
|---|---|
| Sterile pyuria | Very common; WBCs without bacterial growth |
| WBC casts | Highly suggestive of AIN (also seen rarely in acute GN, papillary necrosis) |
| WBCs on microscopy (leukocyturia) | Most sensitive finding |
| Mild proteinuria (<1 g/day) | Tubular, non-nephrotic range in most cases |
| Nephrotic-range proteinuria | Suggests co-existing minimal change disease (NSAIDs) |
| Microhematuria | Common; RBC casts are rare (~26% in one series - unusual) |
| Renal tubular epithelial cells and casts | Reflect tubular injury from infiltrating cells (tubulitis) |
| Granular casts | Also seen (hyaline and granular casts in ~86% of ATIN biopsies) |
| Urine eosinophils | Sensitivity 20-30%, specificity 70-90%; PPV only 15-30% - NOT reliable; use has been largely abandoned |
| Normal urinary sediment | Occurs in ~20% of biopsy-proven ATIN - does NOT exclude the diagnosis |
Blood Tests:
- Elevated serum creatinine/BUN
- Peripheral eosinophilia (inconsistent; more common in drug-induced)
- Elevated ESR, CRP (non-specific)
- Elevated IgE (in drug-induced)
- ANA, ANCA, anti-GBM, complement levels (for immune-mediated causes)
- Serum IgG4 (for IgG4-related disease)
Urine Chemistries:
- FENa is not useful for diagnosing ATIN - values can be above or below 1%; it does not distinguish ATIN from ATN reliably
- Low-grade tubular proteinuria and β2-microglobulinuria indicate proximal tubular disease
Novel Biomarkers (research stage):
- Urine TNF-α and IL-9 - promising for distinguishing AIN from ATN (JCI Insight, 2019)
- Urine MCP-1, NGAL, α1-microglobulin, NAG - elevated in ATIN but not yet validated for clinical discrimination
When to Perform Kidney Biopsy:
- Clinical setting and history do not clearly support ATN or volume depletion
- Tissue diagnosis needed to determine type of lesion, extent of involvement, or degree of fibrosis
- Patient is stable enough for biopsy and potential immunosuppressive therapy
- Choice or duration of therapy is partially determined by biopsy findings
- Goldman-Cecil Medicine, p. 3556-3559
8. Differential Diagnosis
| Feature | AIN | ATN | Prerenal | Glomerulonephritis |
|---|---|---|---|---|
| Urine sediment | WBC casts, pyuria | Muddy brown granular casts | Bland, hyaline casts | RBC casts |
| Proteinuria | <1 g/day (tubular) | Minimal | Minimal | >1 g/day (glomerular) |
| FENa | Variable (not helpful) | >1-2% | <1% | Variable |
| Drug exposure | Common | Common (nephrotoxins) | - | Uncommon |
| Rash/fever/eosinophilia | Variable | No | No | Rare |
| Blood pressure | Usually normal | Normal-elevated | Low | Often elevated |
| Complement | Normal | Normal | Normal | Low (MPGN, lupus) |
9. Management
Step 1: Remove the Offending Cause
The most important first step in drug-induced ATIN is prompt discontinuation of the culprit drug. Kidney function often improves within days to weeks after withdrawal. Delay in drug removal is associated with worse renal recovery and risk of CKD.
Step 2: Treat Underlying Condition
- For infection-related ATIN: treat the primary infection
- For sarcoidosis, SLE, Sjögren: see below
Step 3: Corticosteroids
Evidence base: Small observational studies only - no large RCTs exist. Results are discordant. Early use (within 7-14 days of drug discontinuation, before significant renal damage) may improve recovery. Later use shows less benefit.
Potential regimen (when biopsy confirms ATIN, no contraindications):
- Methylprednisolone IV 250-500 mg/day × 3-4 days, then
- Oral prednisone 1 mg/kg/day, tapered over 8-12 weeks
Corticosteroids are beneficial in:
- Sarcoidosis-related ATIN (good evidence)
- TINU syndrome (3-6 months with slow taper)
- IgG4-related kidney disease (excellent response)
- Drug-induced ATIN if no recovery after drug withdrawal (on case-by-case basis)
Important: Given potential serious side effects of steroids, their use must be individualized. Patient must be stable enough to receive immunosuppression.
Step 4: Alternative Immunosuppression (steroid-resistant/dependent)
- Mycophenolate mofetil (MMF) - used in steroid-intolerant patients; small case series show improvement/stabilization in drug-induced AIN; used in Sjögren and SLE-related ATIN
- Azathioprine, cyclophosphamide - used in Sjögren, ANCA vasculitis-related ATIN
- Methotrexate, cyclosporine - steroid-sparing options in TINU
Step 5: Supportive Care
- Standard AKI management: fluid/electrolyte balance, manage hyperkalemia/acidosis
- Renal replacement therapy (RRT) if severe AKI, refractory complications, or uremia
- Drug dose adjustment for renally cleared medications
Immune Checkpoint Inhibitor-Induced AIN (Emerging Issue)
-
Occurs in ~3% of patients on PD-1/PD-L1/CTLA-4 inhibitors
-
Average latency ~4 months after starting therapy
-
Management: hold checkpoint inhibitor, administer corticosteroids; patients who recover renal function may often safely resume checkpoint inhibitor therapy
-
PPIs may increase risk of AIN in patients already on checkpoint inhibitors
-
Brenner and Rector's The Kidney, p. 1271
-
Harrison's 22E, p. 1461
10. Prognosis
- Majority recover renal function if the offending cause is removed promptly and the disease is recognized early
- Incomplete recovery and progression to CKD is common when:
- Diagnosis is delayed (sustained injury → fibrosis)
- Significant interstitial fibrosis present on biopsy at diagnosis
- Older patients with underlying CKD
- The degree of interstitial fibrosis and tubular atrophy on biopsy is the strongest histologic predictor of long-term renal outcome
- Rifampin-induced ATIN has a particularly severe course (dialysis in ~2/3 of patients)
- TINU syndrome generally has a relatively favorable prognosis with treatment, though uveitis may relapse
11. Special Entities Summary
| Condition | Key Distinguishing Feature | Treatment |
|---|---|---|
| PPI-induced ATIN | Long latency (weeks-months), few allergic features, all PPIs implicated | Discontinue PPI, consider steroids |
| NSAID-induced ATIN | Long latency, minimal allergic features, may have minimal-change co-existing, eosinophils often absent | Discontinue NSAID |
| Rifampin ATIN | Dose-dependent, anti-rifampin antibodies, hemolytic anemia/thrombocytopenia | Discontinue rifampin; frequently requires dialysis |
| Sarcoid ATIN | Noncaseating granulomas, hypercalcemia, hypercalciuria | Steroids (mainstay) |
| TB ATIN | Caseating granulomas, first appearing near glomeruli (high O₂ tension cortex) | Anti-TB therapy |
| TINU syndrome | Adolescent girls, uveitis + ATIN, Fanconi syndrome possible | Steroids 3-6 months |
| IgG4-related | Elevated serum IgG4, storiform fibrosis, multisystem | Steroids (excellent response) |
| Checkpoint inhibitor | ~3% incidence, 4-month latency, may resume drug after recovery | Hold drug + steroids |
Sources:
- Brenner and Rector's The Kidney (2-Volume Set), p. 1595-1606
- Harrison's Principles of Internal Medicine 22E (2025), p. 1457-1473
- Comprehensive Clinical Nephrology 7e, p. 889-895
- Goldman-Cecil Medicine International Edition, p. 3539-3568
- NKF Primer on Kidney Diseases 8e, p. 1432-1436
- Frameworks for Internal Medicine, p. 454-456
Acute tubular injury
Finding Sources
Searching PubMed
"acute tubular injury" AND "acute kidney injury"
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Reading File
I now have comprehensive content from multiple authoritative sources. Let me compile the full answer.
Acute Tubular Injury (ATI)
1. Definition and Terminology
Acute Tubular Injury (ATI) describes damage to renal tubular epithelial cells resulting in a sudden decline in kidney function. It is now the preferred term over the older "acute tubular necrosis (ATN)" because frank, widespread necrosis is rarely seen on histology - injury may be limited to highly susceptible zones, or may manifest as apoptosis and sublethal cellular dysfunction rather than true necrosis. ATI is the most common cause of acute kidney injury (AKI) in hospitalized patients.
Key features:
-
Sudden GFR decline with elevated serum creatinine
-
May produce oliguria (urine output <400 mL/day) or be non-oliguric
-
Shedding of granular casts and tubular epithelial cells into urine
-
Generally reversible if the precipitating cause is corrected
-
Robbins & Kumar Basic Pathology, p. 519
-
NKF Primer on Kidney Diseases 8e, p. 972-976
2. Why the Proximal Tubule is Most Vulnerable
The proximal tubule - especially the S3 segment (straight portion) in the outer medullary stripe - bears the brunt of ATI for several reasons:
| Factor | Explanation |
|---|---|
| High metabolic demand | Packed with mitochondria; dependent exclusively on oxidative phosphorylation (minimal anaerobic glycolysis capacity) |
| High O₂ consumption | Required to generate ATP for active transport and reabsorption |
| Regional blood flow | Outer medulla receives primarily venous capillary flow - persists ischemic even when cortical flow recovers |
| Toxin concentration | High rate of endocytosis; active transport of organic acids and ions concentrates nephrotoxins intracellularly |
| Large reabsorptive surface area | Maximizes exposure of epithelial cells to luminal solutes and toxins |
| Luminal solute concentration | Water reabsorption progressively concentrates toxins in the tubular lumen |
- Robbins & Kumar Basic Pathology, p. 519
- NKF Primer on Kidney Diseases 8e, p. 974
3. Forms and Causes
A. Ischemic ATI (most common)
Results from inadequate renal blood flow - either systemic or localized.
Hypotension-induced (systemic):
| Clinical Scenario | Mechanism |
|---|---|
| Septic shock | Hypoperfusion + inflammatory cytokines (microvascular dysfunction, glycocalyx disruption, mitochondrial dysfunction) |
| Cardiogenic shock | Reduced cardiac output + venous congestion |
| Hypovolemic shock | Hemorrhage, burns, severe diarrhea, third-spacing |
| Acute decompensated heart failure | Both low forward flow and elevated venous pressure reduce GFR |
| Major surgery / trauma | Hypotension + blood loss |
| Autonomic dysfunction | Impaired vasomotor control |
Sepsis creates a particularly hostile environment: beyond hypoperfusion, inflammatory cytokines independently cause variation in renal macro- and microcirculation, diffusion limitation, and mitochondrial dysfunction - injury may occur even without overt hypotension.
Localized renal vascular obstruction:
- Cholesterol atheroembolic disease
- Malignant hypertension
- Small vessel vasculitis
- Thrombotic microangiopathy (TMA)
- Renal artery stenosis / thrombosis
- Renal vein thrombosis
Pigment nephropathy (ischemic + toxic combination):
- Mismatched blood transfusions / hemolytic crises → hemoglobinuria
- Rhabdomyolysis → myoglobinuria
- (Detailed below)
B. Nephrotoxic ATI
Exogenous nephrotoxins:
| Category | Agents |
|---|---|
| Antibiotics | Aminoglycosides (gentamicin, tobramycin), vancomycin, polymyxins, amphotericin B |
| Chemotherapy | Cisplatin, ifosfamide, pemetrexed |
| Contrast agents | Iodinated radiocontrast |
| Immunosuppressants | Cyclosporine, tacrolimus (vasoconstriction) |
| Antivirals | Tenofovir, foscarnet, acyclovir (intratubular crystallization) |
| Analgesics | NSAIDs (prostaglandin-dependent afferent vasodilation impaired) |
| Heavy metals | Mercury, lead, cadmium, arsenic |
| Organic solvents | Ethylene glycol (→ calcium oxalate crystals), carbon tetrachloride |
Endogenous nephrotoxins:
-
Myoglobin (rhabdomyolysis)
-
Hemoglobin (intravascular hemolysis)
-
Uric acid (tumor lysis syndrome → intratubular crystallization)
-
Light chains (multiple myeloma)
-
Calcium oxalate (hyperoxaluria - including from high-dose vitamin C)
-
Robbins & Kumar Basic Pathology, p. 519
-
NKF Primer on Kidney Diseases 8e, p. 988-1000
4. Pathophysiology
The Four Converging Mechanisms of GFR Decline
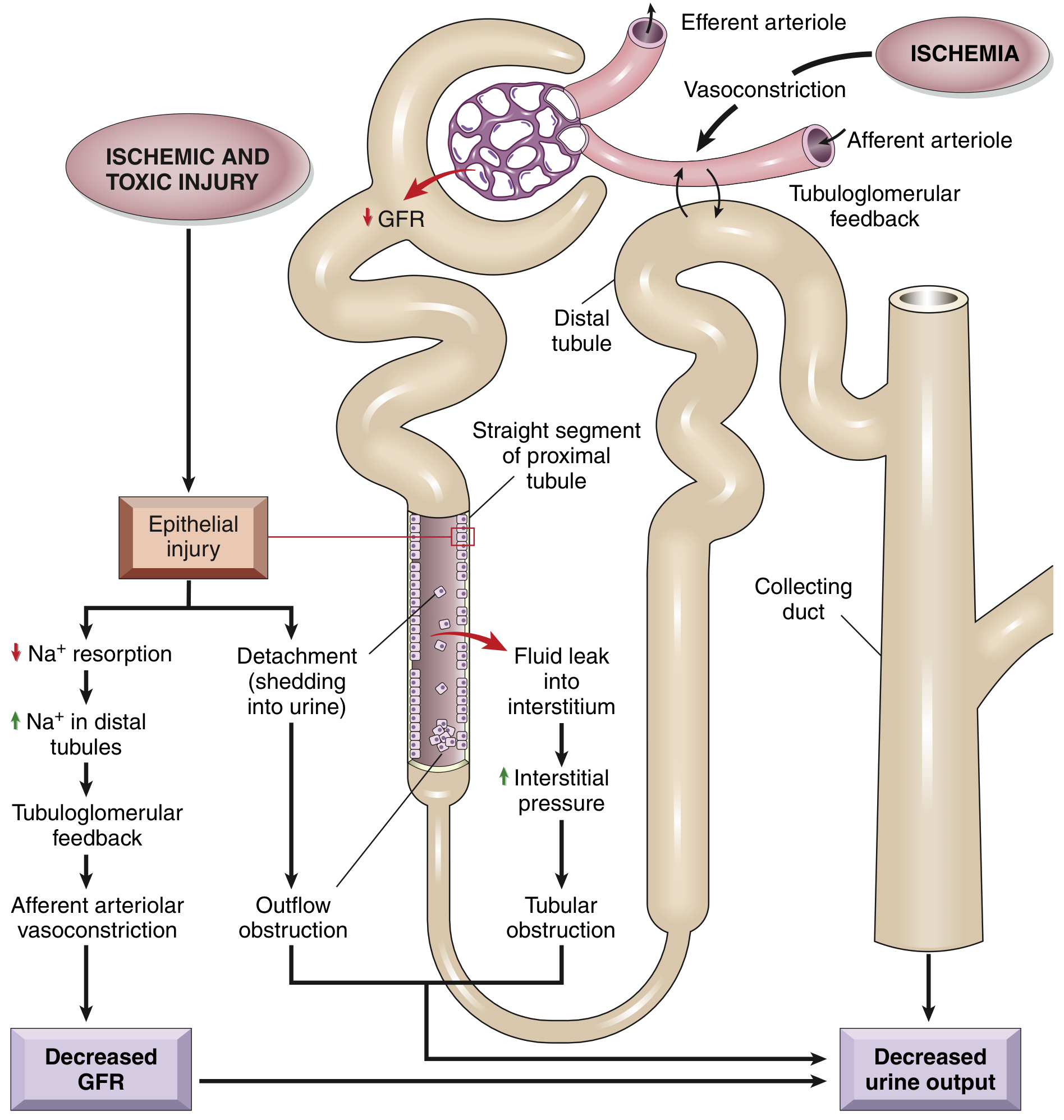
1. Tubuloglomerular Feedback → Afferent Vasoconstriction
- Injured proximal tubular cells fail to reabsorb Na⁺ → increased NaCl delivery to macula densa → tubuloglomerular feedback → afferent arteriolar vasoconstriction via the renin-angiotensin pathway → reduced GFR and O₂ delivery to the outer medulla → further ischemic tubular injury (self-perpetuating cycle)
2. Tubular Cast Obstruction
- Necrotic/apoptotic cells detach from the tubular basement membrane and shed into the lumen
- Detached cells + Tamm-Horsfall protein + plasma proteins form granular casts in distal tubules and collecting ducts
- Casts obstruct outflow → increased intratubular pressure → backpressure against glomerular filtration → further GFR decline
3. Tubular Backleak
- Areas of denuded basement membrane allow glomerular filtrate to leak back into the renal interstitium
- Increases interstitial edema and pressure
- Interstitial pressure compresses peritubular capillaries → perpetuates ischemia
4. Vascular Dysfunction / Hemodynamic Disturbances
-
Decreased vasodilatory prostaglandins and nitric oxide
-
Increased endothelin, angiotensin II, vasopressin
-
Endothelial swelling + leukocyte adhesion in peritubular capillaries (especially vasa recta)
-
Persists even after cortical blood flow normalizes ("extension phase")
-
Robbins & Kumar Basic Pathology, p. 519-521
5. Special Forms: Pigment Nephropathy
Rhabdomyolysis
Breakdown of striated skeletal muscle releasing myoglobin into the circulation.
Causes:
| Category | Examples |
|---|---|
| Physical injury | Trauma, crush injury, compartment syndrome, immobilization |
| Muscle exhaustion | Extreme exercise, seizures, heat stroke, neuroleptic malignant syndrome, malignant hyperthermia |
| Drugs/toxins | Statins, antipsychotics, SSRIs, cocaine, alcohol, amphetamines, heroin |
| Inflammatory | Dermatomyositis, polymyositis, viral infections (influenza, HIV, EBV) |
| Metabolic enzyme defects | McArdle disease (myophosphorylase deficiency), carnitine palmitoyl transferase deficiency |
| Electrolyte disturbances | Severe hypokalemia, hypophosphatemia |
Diagnosis:
- Muscle pain, weakness, dark (cola-colored) urine
- Serum CK >5000 U/L (risk of AKI typically significant when >15,000-20,000 U/L)
- Urine dipstick heme-positive but few/no RBCs on microscopy (myoglobin triggers peroxidase reaction)
- Red-brown pigmented granular casts on urine microscopy
- FENa <1% early (despite intrinsic AKI - due to volume depletion and intense renal vasoconstriction)
- Hyperkalemia, hyperphosphatemia, hyperuricemia, hypocalcemia (early; hypercalcemia may occur during recovery), elevated LDH
Mechanism of tubular injury:
- Myoglobin → renal vasoconstriction (scavenges NO, elevates endothelin and isoprostanes)
- Tubular cast formation: myoglobin + Tamm-Horsfall protein (especially in acidic urine) → tubular obstruction
- Direct tubular cell injury via free radical formation and lipid peroxidation after prolonged heme protein exposure
- NLRP3 inflammasome activation → IL-1β and IL-18 release → proinflammatory cascade
Risk Stratification - McMahon Rhabdomyolysis Risk Score:
| Variable | Score |
|---|---|
| Age >50-70 yrs | 1.5 |
| Age >70-80 yrs | 2.5 |
| Age >80 yrs | 3 |
| Female sex | 1 |
| Creatinine 1.4-2.2 mg/dL | 1.5 |
| Creatinine >2.2 mg/dL | 3 |
| Ca <7.5 mg/dL | 2 |
| CK >40,000 U/L | 2 |
| Non-benign etiology* | 3 |
| Phosphate 4.0-5.4 mg/dL | 1.5 |
| Phosphate >5.4 mg/dL | 3 |
| Bicarbonate <19 mEq/L | 2 |
*Benign = seizures, syncope, exercise, statins, myositis
- Score <5: ~2.3% risk of KRT or death
- Score >10: ~61.2% risk of KRT or death
Treatment:
- Aggressive IV fluid resuscitation: isotonic normal saline targeting urine output >200 mL/hr
- Identify and treat underlying cause; prevent further muscle damage
- Electrolyte correction (hyperkalemia priority)
- Urinary alkalinization (sodium bicarbonate) is theoretically beneficial to reduce cast formation but no clear RCT evidence of superiority
Hemoglobinuria (Intravascular Hemolysis)
Causes: Mismatched blood transfusion, autoimmune hemolytic anemia, G6PD deficiency, mechanical hemolysis (heart valves), malaria, paroxysmal nocturnal hemoglobinuria
Mechanism: Plasma hemoglobin exceeds haptoglobin binding capacity → free Hb dimers filtered at glomerulus → endocytosed by proximal tubular cells or form heme-pigment casts → same four mechanisms as myoglobin (vasoconstriction, cast obstruction, oxidative stress, inflammasome activation)
Treatment: IV fluids (similar to rhabdomyolysis); treat underlying cause
- NKF Primer on Kidney Diseases 8e, p. 1073-1295
6. Morphology / Histology
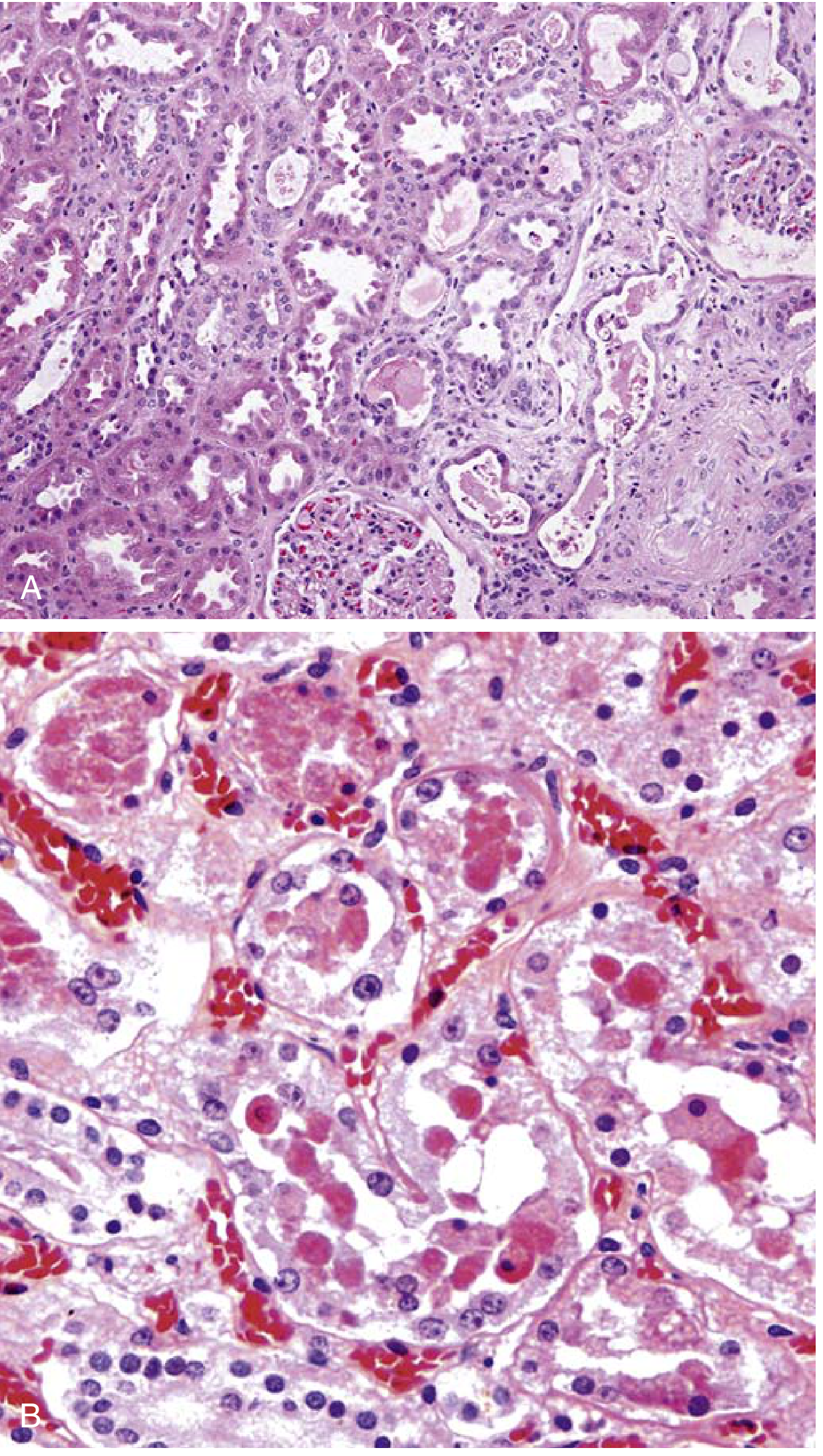
Key Histologic Features:
Ischemic ATI:
- Loss/attenuation of proximal tubular brush border (earliest change)
- Blebbing and sloughing of brush border microvilli
- Vacuolization of tubular cells
- Detachment and sloughing of epithelial cells from basement membranes into the lumen
- Skip lesions: patchy necrosis with large areas of spared tubule between foci - characteristic
- Primary sites: straight segment of proximal tubule (S3) and thick ascending limb of Henle in the outer medulla
- Proteinaceous/granular casts in distal tubules and collecting ducts (Tamm-Horsfall protein + plasma proteins)
- Interstitial edema with mild lymphocytic/neutrophilic/plasma cell infiltrate
- Leukocyte accumulations within dilated vasa recta
- Regenerative changes: flattened epithelium, hyperchromatic nuclei, mitotic figures
Nephrotoxic ATI:
- Histologically similar but more overt necrosis in the proximal convoluted tubule (PCT, S1/S2 segments)
- More continuous (less skip areas) than ischemic ATI
- Agent-specific changes:
- Mercury chloride: Large acidophilic inclusions → necrosis → calcification
- Ethylene glycol: Marked ballooning/hydropic degeneration of PCT + calcium oxalate crystals in tubular lumens
- Myoglobinuria/hemoglobinuria: Red-brown pigmented casts
Regeneration:
- Regenerating cells: flattened, mitotically active, hyperchromatic
- If basement membrane integrity is maintained → re-epithelialization and recovery of function possible
- Robbins & Kumar Basic Pathology, p. 519-521
7. Clinical Features and Phases
The classic course of ATI follows three phases (though many cases, especially nephrotoxic ATI, are non-oliguric):
| Phase | Duration | Key Findings |
|---|---|---|
| Initiation | Hours to ~36 hrs | Dominated by the precipitating event; modest oliguria; modest BUN/Cr rise; may be indistinguishable from prerenal AKI |
| Maintenance (oliguric) | Days to weeks | Oliguria (40-400 mL/day); progressive azotemia; hyperkalemia; metabolic acidosis; fluid/salt overload; uremia; risk of death highest |
| Recovery (polyuric) | Gradual | Diuresis (urine output may reach 3+ L/day as GFR recovers before tubular concentrating ability); risk of hypokalemia, hyponatremia, volume depletion during this phase |
~50% of ATI is non-oliguric - this carries a better prognosis. It is especially common with nephrotoxic ATI (contrast, aminoglycosides).
8. Diagnosis
Urine Microscopy (the "liquid biopsy")
| Finding | Significance |
|---|---|
| "Muddy brown" granular casts | Pathognomonic for ATI; formed from shed tubular epithelial cells + Tamm-Horsfall protein |
| ≥6 granular casts per HPF | LR 10 for ATI; LR 0.10 for prerenal |
| Renal tubular epithelial cells | Shed by the injured tubular epithelium |
| Waxy casts | Represent very dense granular casts; advanced ATI |
| Red-brown pigmented casts | Myoglobinuria/hemoglobinuria |
| Calcium oxalate crystals | Ethylene glycol poisoning |
Urine Chemistry Indices
| Index | ATI | Prerenal |
|---|---|---|
| FENa | >1-2% | <1% |
| FEUrea (on diuretics) | >35-50% | <35% |
| Urine Na | >40 mEq/L | <20 mEq/L |
| Urine osmolality | ~300 mOsm/kg (isosthenuria) | >500 mOsm/kg |
| Specific gravity | ~1.010 | >1.020 |
| BUN:Cr ratio | ~10-15:1 | >20:1 |
Exceptions: FENa may be <1% in ATI caused by radiocontrast, myoglobinuria, or early obstruction due to concurrent intense vasoconstriction.
Novel Biomarkers of Tubular Injury
These can detect tubular injury before creatinine rises, enabling earlier intervention:
| Biomarker | Source | Notes |
|---|---|---|
| NGAL (neutrophil gelatinase-associated lipocalin) | Urine and serum | Rises within 2 hours of ischemia; highly sensitive |
| KIM-1 (kidney injury molecule-1) | Urine | Proximal tubular injury marker |
| IL-18 | Urine | Also upregulated in ischemic ATI |
| TIMP-2 × IGFBP7 (NephroCheck) | Urine | G1 cell-cycle arrest markers; FDA-cleared for risk stratification |
| L-FABP (liver-type fatty acid binding protein) | Urine | Proximal tubule stress marker |
| Cystatin C | Serum | More sensitive than creatinine for early GFR decline |
9. Special Context: COVID-19-Related ATI
AKI occurs in 3-75% of hospitalized COVID-19 patients (depending on disease severity). The most prominent biopsy finding is ATI, though other patterns (glomerular, TMA) also occur.
Mechanisms are multifactorial:
- Critical illness: hypotension, reduced cardiac output, organ cross-talk from ARDS
- Nephrotoxin exposure: drugs used in ICU management
- Positive pressure ventilation: reduces cardiac output and renal perfusion
- Endogenous tubular obstruction: rhabdomyolysis, hyperoxaluria (from IV vitamin C), hyperuricemia
- Direct viral toxicity (controversial): ACE2 receptor on tubular cells serves as viral entry domain; viral RNA detected in urine but not consistently in tissue
- NKF Primer on Kidney Diseases 8e, p. 1297-1307
10. Management
Principles
ATI has no specific pharmacologic therapy proven effective in altering its course. Management is supportive.
1. Remove/Treat the Cause
- Restore hemodynamics and adequate renal perfusion (volume resuscitation, vasopressors for shock)
- Discontinue nephrotoxic drugs
- Treat sepsis (antibiotics, source control)
- Treat underlying condition
2. Fluid Management
- Correct volume depletion; avoid fluid overload (associated with increased mortality)
- Target adequate urine output; monitor for pulmonary edema
- During recovery phase: replace electrolytes lost in diuresis
3. Electrolyte and Acid-Base Management
- Hyperkalemia: ECG monitoring; calcium gluconate (membrane stabilization), insulin/glucose, sodium bicarbonate, potassium binders, dialysis if refractory
- Metabolic acidosis: sodium bicarbonate if severe (pH <7.1)
- Hyponatremia/hypernatremia: appropriate fluid adjustment
- Recovery phase: watch for hypokalemia, hyponatremia
4. Nutrition
- Adequate calorie/protein intake; avoid excessive restriction
5. Drug Dose Adjustment
- All renally cleared drugs must be dose-adjusted based on current GFR
6. Renal Replacement Therapy (RRT)
Indicated for:
- Refractory hyperkalemia
- Refractory metabolic acidosis
- Fluid overload unresponsive to diuretics
- Uremic symptoms (pericarditis, encephalopathy, bleeding)
- Continuous RRT preferred in hemodynamically unstable patients
What Does NOT Work (failed in clinical trials):
- Low-dose ("renal dose") dopamine - increases renal resistance in AKI patients
- Loop diuretics (furosemide) - converts oliguria to non-oliguria but does not change outcomes
- Fenoldopam, atrial natriuretic peptide, thyroid hormone, erythropoietin, IGF-1, prostaglandin A1
Special: Rhabdomyolysis/Pigment Nephropathy
- Aggressive IV normal saline to maintain UO >200 mL/hr
- Urinary alkalinization (sodium bicarbonate) - theoretical benefit (prevents cast formation at alkaline pH), but no RCT evidence of superiority over NS
- Loop diuretics + mannitol - used in some protocols but not proven
11. Prognosis
- Generally reversible if precipitating cause is corrected and basement membrane integrity is preserved (allows re-epithelialization)
- Recovery timeline: urine output increases over days; BUN/Cr normalize over 1-3 weeks
- Poor prognostic factors: pre-existing CKD, older age, multiorgan failure, prolonged ATI (leading to fibrosis), delayed treatment
- ~30% of survivors have incomplete recovery → CKD or ESRD
- In ICU-acquired ATI requiring dialysis: hospital mortality 50-80%
- The patchiness of necrosis and maintenance of tubular basement membrane integrity are the structural basis for recovery - distinguishing ATI from the irreversible renal cortical necrosis
Summary Comparison: ATI vs Renal Cortical Necrosis
| Feature | ATI | Renal Cortical Necrosis |
|---|---|---|
| Necrosis extent | Focal/patchy tubular | Diffuse cortical |
| Reversibility | Usually reversible | Usually irreversible |
| Causes | Ischemia, toxins | Septic abortion, placental abruption, DIC, severe TMA |
| CT appearance | Normal / mild edema | Hypodense areas in renal cortex |
| Basement membrane | Preserved in most areas | Destroyed |
| Prognosis | Variable recovery | Often permanent renal failure |
- Brenner and Rector's The Kidney, p. 2161-2162
Sources:
- Robbins & Kumar Basic Pathology, p. 519-521
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 863-865
- NKF Primer on Kidney Diseases 8e, p. 972-1307
- Comprehensive Clinical Nephrology 7e, p. 393-399
- Brenner and Rector's The Kidney (2-Volume Set), p. 1205-1210, 2161-2162
This is a shared conversation. Sign in to Orris to start your own chat.