DIabetes for 2nd BDS students pathology every thing about it
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Finding Sources
I have all the content I need. Now let me compile the comprehensive answer for 2nd BDS students from Robbins Pathology.
Diabetes Mellitus - Complete Pathology Notes for 2nd BDS
Source: Robbins & Kumar Basic Pathology + Robbins & Cotran Pathologic Basis of Disease
1. DEFINITION
Diabetes mellitus (DM) is a group of metabolic disorders characterized by hyperglycemia, resulting from defects in insulin secretion, insulin action, or most commonly, both. Chronic hyperglycemia causes damage to the kidneys, eyes, nerves, and blood vessels.
2. EPIDEMIOLOGY
- Over 37 million Americans (11% of population) have diabetes
- ~1.4 million new cases diagnosed per year in the USA
- Leading cause of: end-stage renal disease, adult-onset blindness, nontraumatic lower-limb amputations
3. CLASSIFICATION
| Type | Old Name | Key Feature |
|---|---|---|
| Type 1 DM (T1D) | Juvenile-onset / Insulin-dependent | Absolute insulin deficiency (autoimmune destruction of β-cells) |
| Type 2 DM (T2D) | Adult-onset / Non-insulin-dependent | Insulin resistance + relative insulin deficiency |
| Gestational DM | - | Onset during pregnancy |
| Monogenic / Secondary DM | MODY, etc. | Genetic defects, pancreatitis, drugs |
4. DIAGNOSIS (ADA / WHO Criteria)
| Test | Diabetes | Prediabetes |
|---|---|---|
| Fasting plasma glucose | ≥126 mg/dL | 100-125 mg/dL |
| Random plasma glucose | ≥200 mg/dL (with symptoms) | - |
| 2-hr OGTT | ≥200 mg/dL | 140-199 mg/dL |
| HbA1c | ≥6.5% | 5.7-6.4% |
Note: All tests (except symptomatic random glucose) must be confirmed on a separate day.
5. TYPE 1 DIABETES (T1D)
Pathogenesis
T1D is an autoimmune disease resulting in the destruction of pancreatic β-cells. Key steps:
- Genetic predisposition: ~90% of T1D patients carry HLA-DR3 and/or HLA-DR4 alleles. Polymorphisms in the insulin gene promoter also predispose.
- Environmental triggers: Viral infections (Coxsackievirus B, CMV) may initiate autoimmunity by:
- Directly infecting and destroying β-cells
- Inducing an immune response that cross-reacts with β-cell antigens (molecular mimicry)
- Autoimmune destruction: CD4+ and CD8+ T lymphocytes infiltrate the islets (insulitis). Autoantibodies to:
- Glutamic acid decarboxylase (GAD)
- Insulin
- Islet cell antigens (IA-2)
- Progressive loss of β-cells → absolute insulin deficiency
Morphology (Microscopic)
- Insulitis: Lymphocytic infiltration of the islets of Langerhans
- Reduction in islet size and number
- Selective loss of β-cells (α-cells remain intact)
- In end-stage disease: islets appear fibrosed and atrophic
6. TYPE 2 DIABETES (T2D)
Pathogenesis - Two Key Defects
A. Peripheral Insulin Resistance
- Cells (especially muscle, liver, adipose tissue) fail to respond to insulin
- Obesity (especially abdominal/visceral fat) is the major risk factor
- Adipokines (from fat cells) and free fatty acids impair insulin signaling
- Insulin receptors and post-receptor signaling are defective
B. β-Cell Dysfunction (Relative Insulin Deficiency)
- Initially: β-cells compensate by secreting more insulin (hyperinsulinemia)
- Over time: β-cells become exhausted; insulin secretion declines
- Amyloid deposition in islets further impairs function
Role of Obesity
Obesity (especially central/visceral fat) leads to:
- Elevated free fatty acids → lipotoxicity of β-cells
- Adipokines (leptin, adiponectin resistance, resistin) promoting insulin resistance
- Chronic low-grade inflammation (TNF-α, IL-6 from adipose tissue)
Morphology (Microscopic)
- Amyloid (islet amyloid polypeptide / amylin) deposits in the islets - most characteristic finding
- Reduction in number and size of islets (less dramatic than T1D)
- No insulitis (no lymphocytic infiltration)
- β-cell mass is reduced (by ~20-65%)
7. NORMAL INSULIN PHYSIOLOGY (Review)
- Produced by β-cells of the islets of Langerhans (pancreas)
- Stimulated by: glucose, amino acids, GLP-1, GIP
- Key effects:
- Liver: Promotes glycogen synthesis; inhibits gluconeogenesis
- Muscle: Promotes glucose uptake (via GLUT-4) and glycogen synthesis
- Adipose: Promotes lipogenesis; inhibits lipolysis
- All tissues: Promotes protein synthesis
8. MORPHOLOGY OF DIABETES (Summary Table)
| Organ/Tissue | Findings |
|---|---|
| Pancreas - T1D | Insulitis; reduced islets; β-cell loss; leukocyte infiltration |
| Pancreas - T2D | Islet amyloid deposits (amylin); slight reduction in β-cells |
| Blood vessels (macro) | Accelerated atherosclerosis |
| Small vessels (micro) | Microangiopathy - basement membrane thickening |
| Kidney | Diffuse/nodular glomerulosclerosis (Kimmelstiel-Wilson lesion); tubular glycogen (Armanni-Ebstein lesion); arteriolar hyalinosis |
| Eye | Retinal microaneurysms, neovascularization, exudates; cataracts; glaucoma |
| Nerves | Segmental demyelination; Schwann cell injury |
| Skin | Carbuncles, necrobiosis lipoidica diabeticorum |
9. LONG-TERM (CHRONIC) COMPLICATIONS
The diagram below shows the systemic complications of diabetes:
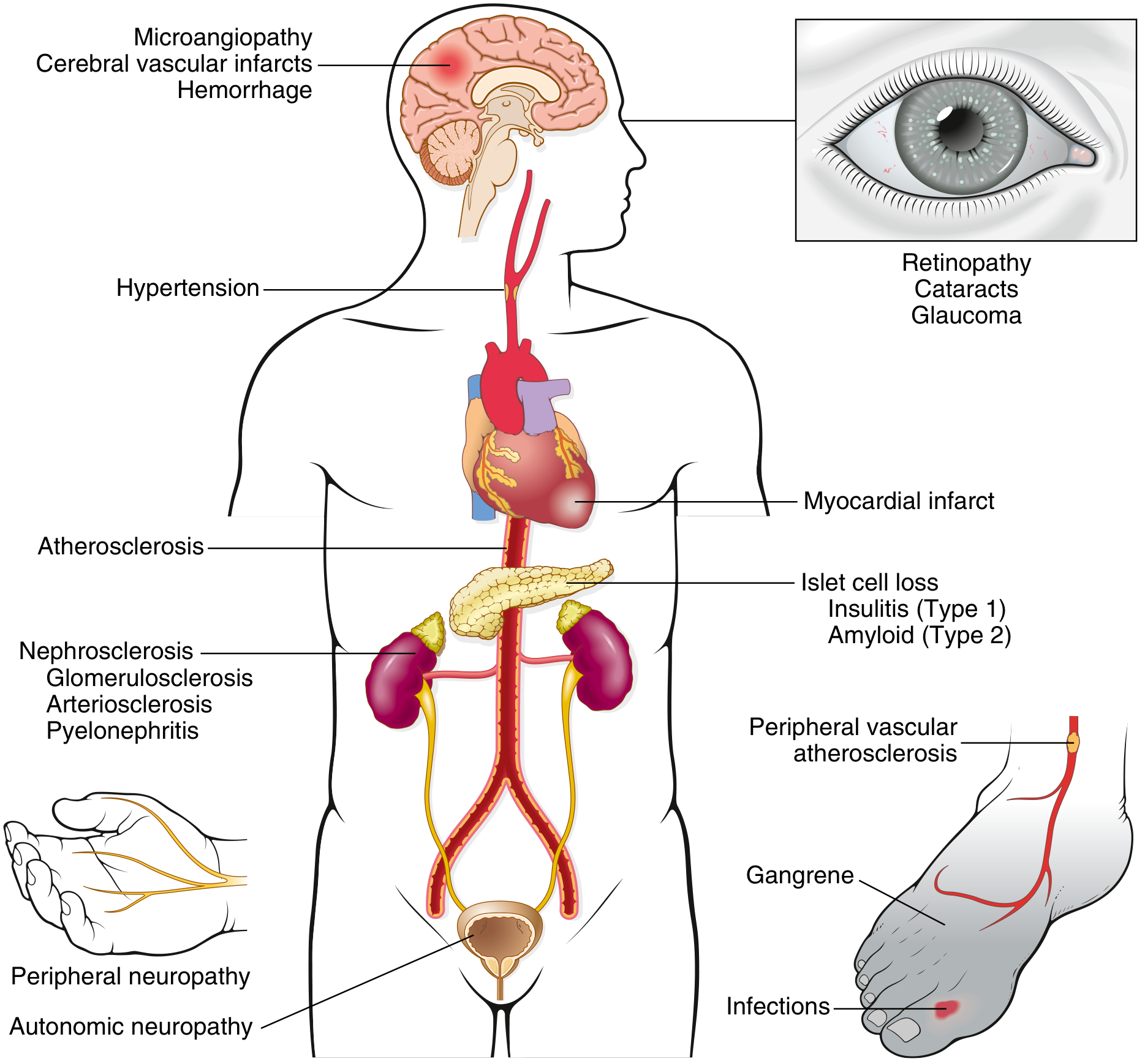
A. Diabetic Macrovascular Disease
- Mechanism: Accelerated atherosclerosis
- Consequences:
- Myocardial infarction (most common cause of death)
- Stroke / cerebral vascular infarcts
- Peripheral vascular disease → gangrene of lower limbs
B. Diabetic Microangiopathy
- Mechanism: Basement membrane thickening of capillaries and small vessels
- Caused by AGEs, PKC activation, polyol pathway disturbances
Retinopathy:
- Microaneurysms (earliest lesion), dot-blot hemorrhages
- Hard exudates (lipid deposits)
- Cotton-wool spots (soft exudates = microinfarcts)
- Neovascularization (proliferative retinopathy)
- Leading cause of adult-onset blindness
Nephropathy:
- Diffuse glomerulosclerosis: Diffuse increase in mesangial matrix and BM thickening (most common)
- Nodular glomerulosclerosis (Kimmelstiel-Wilson lesion): Pathognomonic - ovoid/spherical deposits of laminated matrix in mesangium
- Afferent AND efferent arteriolar hyalinosis (efferent hyalinosis is virtually diagnostic of diabetes)
- Armanni-Ebstein lesion: glycogen accumulation in tubular epithelial cells
- Increased susceptibility to pyelonephritis and necrotizing papillitis
Neuropathy:
- Peripheral neuropathy: segmental demyelination, axonal damage
- Autonomic neuropathy: impaired sweating, postural hypotension, bladder dysfunction, impotence, GI dysmotility
C. Mechanisms of Chronic Complications (4 Main Pathways)
1. Advanced Glycation End Products (AGEs)
- Formed by non-enzymatic glycation of proteins
- AGEs bind RAGE receptors → release TGF-β (fibrosis), VEGF (neovascularization), ROS
- Cross-link extracellular matrix → trap LDL → accelerate atherosclerosis
- Trap albumin in capillary walls → basement membrane thickening
2. Protein Kinase C (PKC) Activation
- Intracellular hyperglycemia → ↑DAG → activates PKC
- Downstream: ↑VEGF (retinopathy), ↑TGF-β (fibrosis, basement membrane deposition)
3. Polyol Pathway
- In insulin-independent tissues (lens, nerves, kidney): excess glucose → sorbitol (via aldose reductase)
- Sorbitol accumulates → osmotic damage, ↓myoinositol, ↓Na+/K+ ATPase activity → nerve dysfunction (neuropathy), cataract
4. Hexosamine Pathway
- Excess glucose → glucosamine-6-phosphate → modifies transcription factors
- Leads to abnormal gene expression and cellular dysfunction
10. ACUTE COMPLICATIONS
| Complication | Type | Features |
|---|---|---|
| Diabetic Ketoacidosis (DKA) | T1D mainly | Absolute insulin deficiency → unregulated lipolysis → fatty acids → ketones (β-hydroxybutyrate, acetoacetate) → metabolic acidosis, Kussmaul breathing, fruity breath |
| Hyperosmolar Hyperglycemic State (HHS) | T2D mainly | Extreme hyperglycemia (>600 mg/dL), severe dehydration, no significant ketoacidosis (residual insulin prevents lipolysis) |
| Hypoglycemia | Both types | Excess insulin/oral agents; can cause loss of consciousness |
DKA Mechanism:
- No insulin → ↑glucagon → ↑gluconeogenesis, ↑glycogenolysis
- ↑lipolysis → ↑free fatty acids → ↑ketogenesis in liver
- Ketones = organic acids → metabolic acidosis
- Osmotic diuresis → dehydration, electrolyte loss
11. CLINICAL FEATURES
Classic Triad ("3 Polys")
- Polyuria - osmotic diuresis from glycosuria (glucose exceeds renal threshold ~180 mg/dL)
- Polydipsia - secondary to water loss
- Polyphagia - cellular starvation despite hyperglycemia (cells cannot uptake glucose)
Other Features
- Weight loss (especially T1D - catabolism)
- Fatigue, blurred vision
- Recurrent infections (impaired leukocyte function, high glucose supports microbial growth)
HbA1c (Glycated Hemoglobin)
- Reflects average blood glucose over past 2-3 months (lifespan of RBC)
- Formed by non-enzymatic glycation of Hb-A by glucose
- Target in diabetic management: <7%
- Diagnostic of DM: ≥6.5%
12. GESTATIONAL DIABETES MELLITUS (GDM)
- Glucose intolerance first recognized during pregnancy
- Due to placental hormones (hPL, progesterone, cortisol) causing insulin resistance
- Risk: macrosomia, neonatal hypoglycemia, increased risk of T2D later in both mother and child
- Screening: 24-28 weeks gestation with 50g GCT, confirmed with 75g OGTT
13. MONOGENIC / SECONDARY DIABETES
MODY (Maturity-Onset Diabetes of the Young)
- Autosomal dominant mutations in single genes affecting β-cell function
- MODY 2: Glucokinase gene mutation (glucose sensor defect)
- MODY 3: HNF-1α mutation (most common MODY)
Secondary Causes:
- Pancreatitis / pancreatectomy
- Hemochromatosis (iron deposits destroy islets - "bronze diabetes")
- Cushing's syndrome (↑cortisol → insulin resistance)
- Acromegaly (↑GH → insulin resistance)
- Drug-induced (glucocorticoids, thiazide diuretics, protease inhibitors)
14. ORAL / DENTAL RELEVANCE FOR BDS STUDENTS
Diabetes has important implications for dental practice:
| Oral Feature | Mechanism |
|---|---|
| Periodontal disease (most common oral complication) | Microangiopathy of periodontal vessels + impaired leukocyte function + altered collagen metabolism → severe, rapidly progressing periodontitis |
| Xerostomia (dry mouth) | Autonomic neuropathy of salivary glands; dehydration |
| Increased caries | Reduced salivary flow; high glucose in saliva |
| Oral candidiasis | Immunosuppression + high salivary glucose |
| Poor wound healing | Microangiopathy + impaired neutrophil function + neuropathy |
| Burning mouth syndrome | Peripheral neuropathy |
| Tooth mobility | Alveolar bone loss from severe periodontitis |
| Acetone breath | DKA - recognizable in dental chair as fruity/acetone odor |
Key dental management point: Never perform elective dental procedures if blood glucose is >200 mg/dL. Risk of hypoglycemia during dental visits (stress-induced, missed meals). Always have glucose source available.
15. COMPARISON: T1D vs T2D
| Feature | Type 1 DM | Type 2 DM |
|---|---|---|
| Age of onset | Usually <30 yrs (juvenile) | Usually >40 yrs (but rising in youth) |
| Body habitus | Normal / thin | Obese (80%) |
| Onset | Abrupt | Insidious |
| Insulin levels | Very low / absent | Normal or elevated initially |
| C-peptide | Absent | Present |
| Autoantibodies | Present (anti-GAD, anti-insulin) | Absent |
| HLA association | HLA-DR3, DR4 | Weak HLA association |
| Pathology | Insulitis, β-cell destruction | Islet amyloid, insulin resistance |
| Ketoacidosis | Common | Rare |
| Treatment | Insulin mandatory | Diet, exercise, oral agents, ± insulin |
16. QUICK EXAM BUZZWORDS
- Kimmelstiel-Wilson nodules = nodular glomerulosclerosis in T2D (pathognomonic)
- Insulitis = lymphocytic infiltration of islets = T1D
- Islet amyloid (amylin/IAPP deposits) = T2D
- Armanni-Ebstein lesion = glycogen in tubular cells = uncontrolled DM
- RAGE = receptor for AGEs
- Efferent arteriolar hyalinosis = virtually diagnostic of DM (different from hypertension which only affects afferent)
- HbA1c ≥6.5% = diagnostic
- MODY = Maturity Onset Diabetes of the Young
- DKA = T1D; HHS = T2D
- Sorbitol = polyol pathway → cataract + neuropathy
Sources: Robbins & Kumar Basic Pathology (9780323790185), Robbins & Cotran Pathologic Basis of Disease (9780443264528)
This is a shared conversation. Sign in to Orris to start your own chat.