EXPLAIN Diabetes mallitus 1 and 2
Reading File
Finding Sources
Searching PubMed
"diabetes mellitus" AND "type 1" AND "type 2"
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Searching Images
diabetes mellitus type 1 type 2 comparison pathophysiology diagram
I now have comprehensive information from multiple authoritative textbooks. Let me compile a thorough explanation.
Diabetes Mellitus: Type 1 & Type 2
Diabetes mellitus (DM) is a syndrome of impaired carbohydrate, fat, and protein metabolism caused by either lack of insulin secretion or decreased sensitivity of tissues to insulin. — Guyton and Hall Textbook of Medical Physiology
Overview & Classification
| Feature | Type 1 DM | Type 2 DM |
|---|---|---|
| Former name | Insulin-dependent / Juvenile DM | Non-insulin-dependent DM |
| Core defect | Absolute insulin deficiency | Insulin resistance + relative insulin deficiency |
| Prevalence | ~5–10% of all DM | ~90–95% of all DM |
| Typical onset | Childhood/adolescence (but any age) | Adults >40 (but rising in youth) |
| Body habitus | Usually lean | Often overweight/obese |
| Autoimmunity | Yes (T-cell mediated) | No |
| Ketoacidosis risk | High | Low (except in severe stress) |
| Insulin required | Always | Eventually in many cases |
TYPE 1 DIABETES MELLITUS
Pathogenesis
Type 1 DM results from cellular-mediated autoimmune destruction of insulin-secreting pancreatic β-cells. In the vast majority of patients, destruction is mediated by T cells (termed Type 1A or immune-mediated diabetes). The α-, δ-, and other islet cells are preserved. The islets show a chronic mononuclear cell infiltrate called insulitis. — Tietz Textbook of Laboratory Medicine, 7th Ed.
The autoimmune process begins months to years before clinical presentation, and an 80–90% reduction in β-cell volume is required before symptomatic diabetes appears. The rate of destruction is typically faster in children than adults.
Genetic factors: Strong HLA association (HLA-DR3, HLA-DR4). Up to 40% of individuals develop Type 1 DM after age 30.
Autoantibodies (Markers of β-cell autoimmunity)
The following autoantibodies appear in serum years before hyperglycemia:
- Islet Cell Antibodies (ICAs) — present in 75–85% of newly diagnosed patients
- Insulin Autoantibodies (IAA) — present in ~80–90% of children who develop T1DM before age 5
- Anti-GAD65 (glutamic acid decarboxylase) — highly specific
- Anti-IA-2 (islet antigen-2 / tyrosine phosphatase)
- Anti-ZnT8 (zinc transporter)
Clinical Presentation
Type 1 DM may develop abruptly over days to weeks, with three principal features:
- Increased blood glucose (hyperglycemia)
- Increased fat utilization/ketone production → diabetic ketoacidosis (DKA)
- Depletion of body proteins
Classic "3 Ps": Polyuria, Polydipsia, Polyphagia — plus weight loss and fatigue.
TYPE 2 DIABETES MELLITUS
Pathogenesis
Type 2 DM is initially caused by decreased sensitivity of target tissues to insulin (insulin resistance), particularly in liver, muscle, and adipose tissue. In response, the pancreas hypersecetes insulin. Over time, β-cells become dysfunctional and fail, leading to progressive hyperglycemia. — Guyton and Hall Textbook of Medical Physiology
Key drivers:
- Insulin resistance (muscle, liver, adipose)
- β-cell exhaustion/failure (progressive)
- Excess hepatic glucose production
- Incretin defect (reduced GLP-1 response)
Risk factors include: Obesity (especially visceral adiposity), physical inactivity, family history, age >45, hypertension, dyslipidemia, gestational DM history, polycystic ovary syndrome (PCOS), certain ethnicities. — Textbook of Family Medicine, 9e
Pathophysiology in Detail
With prolonged and severe insulin resistance, even elevated insulin levels cannot maintain normal glucose regulation. In early disease, postprandial hyperglycemia predominates. In later stages, fasting hyperglycemia develops as β-cell dysfunction worsens. Some patients never develop overt DM despite insulin resistance if their pancreas can sustain compensatory hypersecretion; others progress to full-blown DM, especially with genetic predisposition.
DIAGNOSIS
Both types share the same diagnostic criteria:
| Test | Diagnostic Threshold |
|---|---|
| Fasting plasma glucose | ≥ 126 mg/dL (7.0 mmol/L) |
| 2-hr OGTT plasma glucose | ≥ 200 mg/dL (11.1 mmol/L) |
| HbA1c | ≥ 6.5% (48 mmol/mol) |
| Random glucose + symptoms | ≥ 200 mg/dL |
METABOLIC CONSEQUENCES
When insulin is absent or ineffective, the following occur in both types (more severe in T1DM without treatment):
- Hyperglycemia — blood glucose may reach 300–1200 mg/dL in untreated T1DM; renal threshold (~180–200 mg/dL) is exceeded → glucosuria
- Osmotic diuresis → polyuria → dehydration → polydipsia
- Cellular starvation despite hyperglycemia → polyphagia, weight loss
- Increased lipolysis → elevated free fatty acids → ketogenesis → DKA (especially T1DM)
- Protein catabolism → muscle wasting — Guyton and Hall Textbook of Medical Physiology
COMPLICATIONS
Microvascular (Small Vessel Disease)
- Diabetic Retinopathy — leading cause of new blindness in adults; background → proliferative
- Diabetic Nephropathy — leading cause of end-stage renal disease; earliest sign is microalbuminuria
- Diabetic Neuropathy — peripheral sensorimotor + autonomic; glove-and-stocking pattern
Macrovascular (Large Vessel Disease)
- Atherosclerosis accelerated (CAD, stroke, peripheral artery disease)
- Ischemic heart disease — leading cause of mortality in T2DM
- Lower extremity ischemia → diabetic foot ulcers, amputations
The Diabetes Control and Complications Trial (DCCT) demonstrated that tight glycemic control significantly reduces microvascular complications (microalbuminuria, neuropathy, retinopathy). — Rosen's Emergency Medicine
Long-term complications involve both macroangiopathy (atherosclerosis, ischemic heart disease, lower extremity ischemia) and microangiopathy (retinopathy, nephropathy, neuropathy). — Robbins & Cotran Pathologic Basis of Disease
TREATMENT
Type 1 DM
Insulin replacement is mandatory. Goals:
- Mimic physiologic insulin secretion with basal-bolus regimen
- Delivery via: Multiple Daily Injections (MDI), Continuous Subcutaneous Insulin Infusion (CSII/pump), Automated Insulin Delivery (AID) systems with CGM
- Insulin types range from rapid-acting (aspart, lispro; onset <15 min) to ultralong-acting (degludec; duration ~42 hrs) — Harrison's Principles of Internal Medicine, 22E (2025)
Type 2 DM
A progressive, multi-step approach: — Harrison's Principles of Internal Medicine, 22E
- Lifestyle modification first — caloric restriction, exercise, weight loss
- Metformin (first-line): reduces hepatic glucose production, improves insulin sensitivity, promotes modest weight loss, low cost
- Sulfonylureas (glimepiride, glipizide): stimulate insulin secretion via ATP-sensitive K⁺ channels
- GLP-1 receptor agonists (semaglutide, liraglutide): enhance insulin secretion, suppress glucagon, cause significant weight loss
- GIP/GLP-1 dual agonists (tirzepatide): greater weight reduction + glycemic control
- SGLT-2 inhibitors (empagliflozin, dapagliflozin): promote urinary glucose excretion; proven cardiovascular and renal benefits
- DPP-4 inhibitors (sitagliptin): prolong incretin effect
- Thiazolidinediones (pioglitazone): increase insulin sensitivity via PPARγ
- Insulin — often required in later stages
PATHOPHYSIOLOGY DIAGRAM
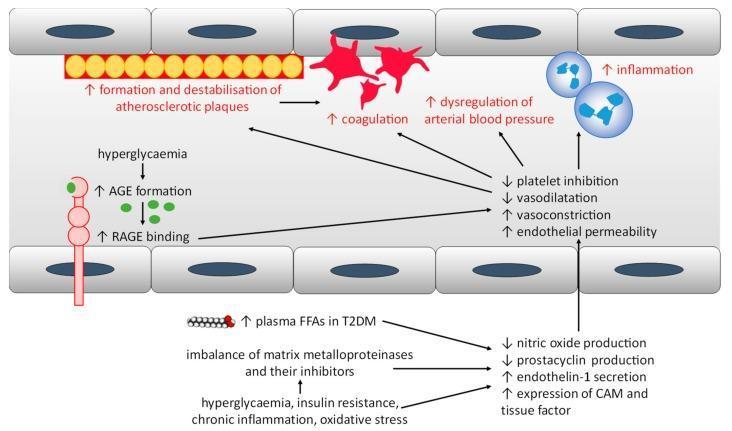
Mechanisms of endothelial dysfunction and increased cardiovascular risk in both T1DM and T2DM — driven by hyperglycemia, AGE formation, oxidative stress, and insulin resistance.
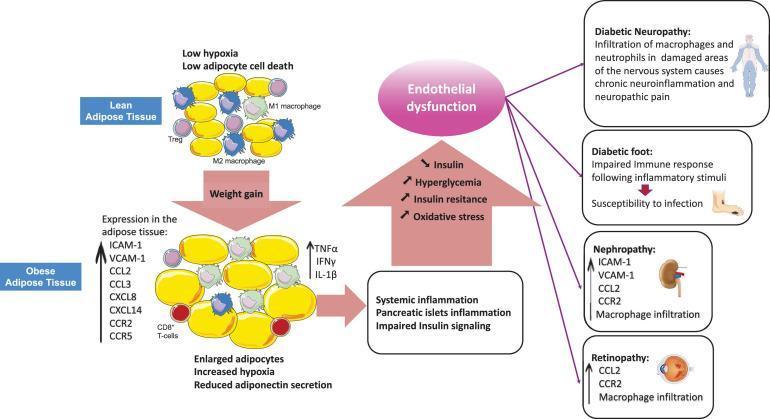
Progression from obesity-induced adipose inflammation → systemic inflammation → β-cell dysfunction → insulin resistance → hyperglycemia → diabetic complications (neuropathy, nephropathy, retinopathy, diabetic foot).
KEY DIFFERENCES SUMMARY
| Type 1 | Type 2 | |
|---|---|---|
| Mechanism | Autoimmune β-cell destruction | Insulin resistance + β-cell failure |
| Insulin levels | Very low/absent | Initially high (compensatory), then falls |
| C-peptide | Absent/very low | Present |
| Autoantibodies | Positive (GAD, IAA, ICA, IA-2) | Negative |
| Ketoacidosis | Common if untreated | Rare |
| HHS | Rare | Common (hyperosmolar hyperglycemic state) |
| Treatment | Insulin always required | Lifestyle → oral agents → insulin |
| Cardiovascular risk | Increased | High (leading cause of death) |
| Associated conditions | Other autoimmune diseases (thyroiditis, celiac, Addison's) | Obesity, HTN, dyslipidemia, PCOS, metabolic syndrome |
Sources: Guyton & Hall Textbook of Medical Physiology | Harrison's Principles of Internal Medicine, 22E (2025) | Tietz Textbook of Laboratory Medicine, 7th Ed. | Robbins & Cotran Pathologic Basis of Disease | Goodman & Gilman's Pharmacological Basis of Therapeutics | Rosen's Emergency Medicine
EXPLAIN Routes of excretion
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Searching Images
routes of drug excretion pharmacokinetics kidney biliary lung diagram
Reading File
I now have comprehensive content from multiple authoritative pharmacology textbooks. Here is the complete explanation:
Routes of Drug Excretion
Excretion is the process by which drugs and their metabolites are irreversibly removed from the body. Drugs are eliminated either unchanged or as metabolites. Excretory organs (the lung excluded) eliminate polar compounds more efficiently than lipid-soluble substances. Thus, lipid-soluble drugs are not readily excreted until they are metabolized to more polar forms via Phase I and Phase II reactions. — Goodman & Gilman's Pharmacological Basis of Therapeutics
Total body clearance is the sum of all routes:
CL_total = CL_renal + CL_hepatic + CL_pulmonary + CL_other
1. RENAL EXCRETION (Most Important Route)
The kidney is the most important organ for excreting drugs and their metabolites. Renal excretion of unchanged drug accounts for 25–30% of drugs administered to humans. — Goodman & Gilman's
Renal excretion involves three distinct processes:
A. Glomerular Filtration
- Drugs enter the kidney through renal arteries → glomerular capillary plexus
- Only free (unbound) drug passes through capillary slits into the Bowman space; protein-bound drug is not filtered
- The GFR is ~120 mL/min/1.73 m² normally; reduced in renal disease
- Lipid solubility and pH do not influence glomerular filtration — only GFR and protein binding matter
B. Active Tubular Secretion (Proximal Tubule)
- Drugs not filtered leave via efferent arterioles → surround the proximal tubular lumen
- Two energy-dependent active transport systems:
- Anion transporters (OAT1, OAT3): for weak acids (e.g., penicillin, furosemide, indomethacin)
- Cation transporters: for weak bases (e.g., amiloride, dopamine, histamine)
- These systems have low specificity → drug competition occurs (e.g., probenecid blocks OAT1/OAT3, inhibiting penicillin secretion → prolonged antibiotic effect)
- Penicillin has t½ = 30 min largely due to rapid tubular secretion
- Neonates and premature infants have incompletely developed tubular secretory mechanisms → drug retention risk
C. Passive Tubular Reabsorption (Distal Tubule — "Ion Trapping")
- As drug concentration rises in the tubule, uncharged drug diffuses back into systemic circulation
- Ionized (charged) drug cannot be reabsorbed → stays in urine → excreted
- Manipulation of urine pH exploits this:
- Alkalinization (e.g., NaHCO₃): ionizes weak acids → increases their excretion (e.g., phenobarbital, salicylate overdose)
- Acidification (e.g., ammonium chloride): ionizes weak bases → increases their excretion (e.g., amphetamine)
- This principle is used clinically in drug poisoning management
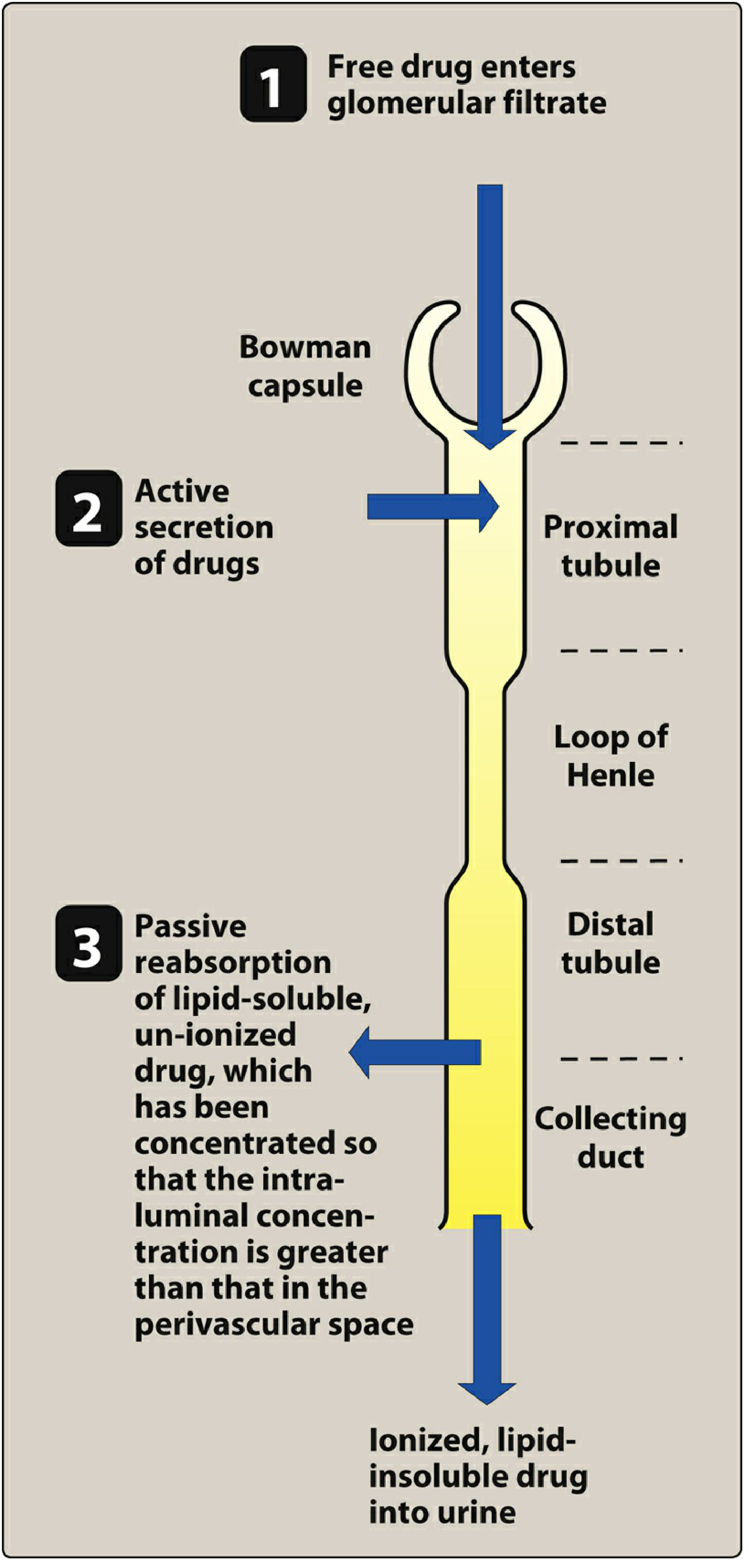
Drug elimination by the kidney: filtration → secretion → reabsorption. — Lippincott's Illustrated Reviews: Pharmacology
Clinical Considerations
- Elderly patients: Renal function declines ~1% per year → increased drug levels and prolonged duration → dose adjustment required
- Renal disease: Drugs primarily excreted by kidney accumulate → toxicity risk
- Creatinine clearance (CrCl) is used to estimate GFR and guide dosing of renally cleared drugs (e.g., methotrexate, aminoglycosides, digoxin)
2. BILIARY / FECAL EXCRETION
Hepatocytes actively secrete drugs and metabolites into bile via canalicular membrane transporters (ABC family: MRP2, P-glycoprotein, BCRP). These excreted substances are then released into the GI tract during digestion. — Goodman & Gilman's
Substances excreted in the feces are:
- Unabsorbed orally ingested drugs
- Drug metabolites excreted in bile
- Drugs secreted directly into the intestinal tract and not reabsorbed
Enterohepatic Recycling (Circulation)
A critical concept:
- Conjugated metabolites (e.g., glucuronides) secreted into bile may be hydrolyzed back by intestinal microflora in the gut
- The parent drug is then reabsorbed → returned to systemic circulation
- If extensive, enterohepatic recycling significantly prolongs drug half-life and effects
- Examples: oral contraceptives (estrogens), morphine glucuronides, digoxin, chloramphenicol
- Interrupting enterohepatic cycling: Give oral agents that bind bile metabolites (e.g., bile acid sequestrants, activated charcoal, cholestyramine) to enhance fecal elimination — useful in drug overdose
3. PULMONARY EXCRETION (Lungs)
The lungs are the primary route for eliminating volatile anesthetic gases (e.g., desflurane, sevoflurane, isoflurane, nitrous oxide). Reversal of CNS effects of inhaled anesthetics depends on pulmonary excretion. — Miller's Anesthesia
Key points:
- Drugs with high volatility and low blood-gas partition coefficient are eliminated faster via the lungs
- Non-volatile products of anesthetic metabolism are eliminated by the kidney
- The lungs also eliminate CO₂ (waste product of metabolism) via ventilation
- Example: ethanol is partially excreted via lungs (basis of breathalyzer tests — breath alcohol correlates well with blood alcohol)
- DMSO (dimethyl sulfoxide) produces garlic-like breath odor from partial pulmonary excretion
4. BREAST MILK EXCRETION
An estimated 50–70% of breastfeeding mothers in the U.S. take some medication, making this route clinically significant. — Goodman & Gilman's
Key principles:
- Breast milk is slightly more acidic than plasma → basic (alkaline) drugs concentrate in milk; acidic drugs are at lower concentration in milk than plasma
- Nonelectrolytes (e.g., ethanol, urea) reach the same concentration in milk as in plasma regardless of pH
- Lipid-soluble drugs, drugs with low protein binding, and low-molecular-weight drugs penetrate milk more readily
- Exposure risk to nursing infant: infants have lower body mass and immature drug-metabolizing capacity → greater susceptibility to drug effects
- Resource for clinicians: LactMed® database (National Library of Medicine)
5. OTHER MINOR ROUTES
| Route | Significance | Notes |
|---|---|---|
| Sweat | Minor | Used forensically for drug detection |
| Saliva | Minor | Cotinine (nicotine metabolite), some drugs detectable |
| Tears | Minor | Quantitatively negligible |
| Hair | Minor | Forensic significance; drugs (e.g., cocaine, heavy metals) incorporated into hair matrix |
| Skin | Minor | Some toxins (heavy metals) secreted |
"Excretion of drugs into sweat, saliva, and tears is quantitatively unimportant... excretion into hair and skin is quantitatively unimportant, but sensitive methods of detection of drugs in these tissues have forensic significance." — Goodman & Gilman's
Summary Table
| Route | Primary Organ | Drugs Eliminated | Clinical Relevance |
|---|---|---|---|
| Renal (urine) | Kidney | Most polar/water-soluble drugs; 25–30% unchanged | Dose adjust in renal failure; ion trapping in overdose |
| Biliary/Fecal | Liver → GI tract | High-MW conjugates, lipophilic drugs | Enterohepatic recycling prolongs drug effects |
| Pulmonary | Lungs | Volatile anesthetics, ethanol, CO₂ | Speed of recovery from inhalational anesthesia |
| Breast milk | Mammary glands | Basic drugs, lipophilic drugs, nonelectrolytes | Risk to nursing infant |
| Sweat/Saliva/Hair | Skin/salivary glands/hair follicles | Trace amounts | Forensic toxicology |
Sources: Goodman & Gilman's Pharmacological Basis of Therapeutics | Lippincott's Illustrated Reviews: Pharmacology | Miller's Anesthesia, 10th Ed. | Katzung's Basic & Clinical Pharmacology, 16th Ed.
This is a shared conversation. Sign in to Orris to start your own chat.