Heme synthesis
Reading File
Finding Sources
Searching the Web
heme synthesis pathway steps enzymes regulation
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have all the information needed for a comprehensive answer. Let me compose the full response.
Heme Synthesis
Structure of Heme
Heme (ferrous protoporphyrin IX) consists of a porphyrin ring coordinated with an Fe²⁺ atom. Four pyrrole rings are joined by methenyl bridges (=CH-) to form the macrocycle. Eight side chains (two per pyrrole) include methyl (M), vinyl (V), and propionate (P) groups arranged in the order M-V / V-M / V-P / P-M - the type III series, the most abundant in nature.
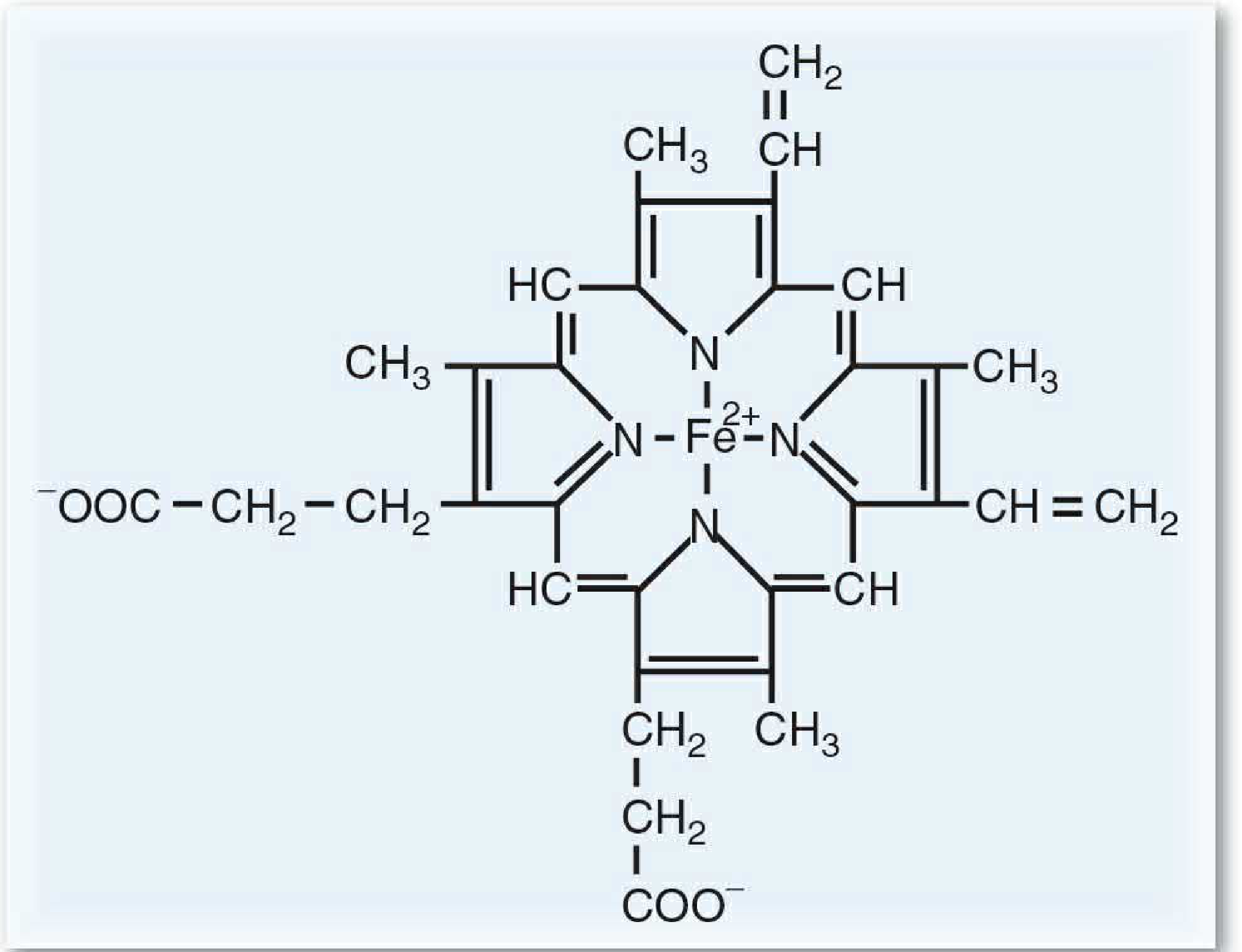
Heme is complexed with proteins to form hemoglobin, myoglobin, and cytochromes (including cytochrome P450).
Location
Heme synthesis takes place partly in the mitochondria and partly in the cytoplasm:
- Mitochondria: steps 1, 6, 7, 8 (first and last three steps)
- Cytoplasm: steps 2-5
The two main sites of synthesis are erythroid precursors in bone marrow (to support hemoglobin) and hepatocytes (to support cytochrome P450 enzymes).
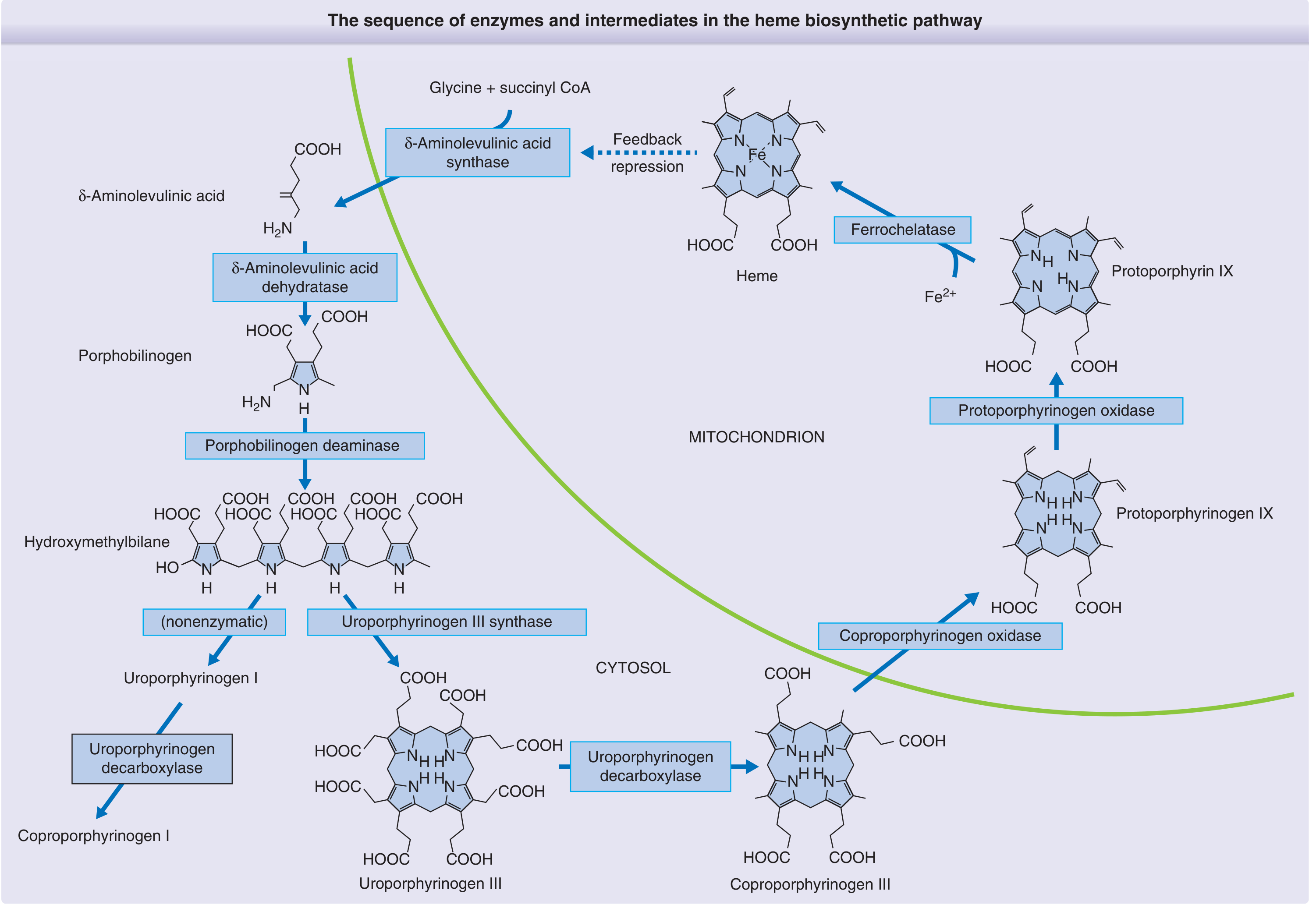
The 8-Step Pathway
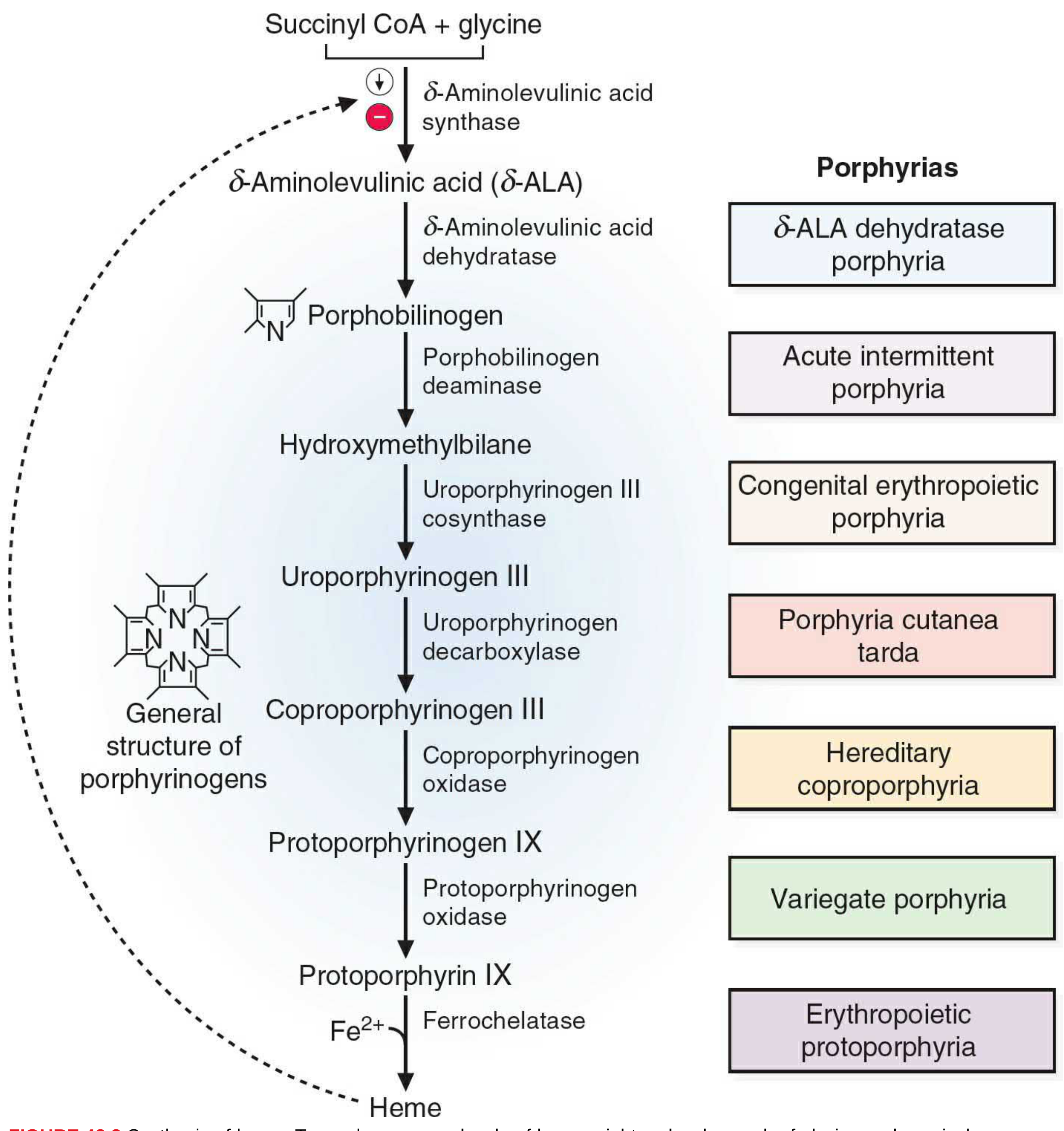
| Step | Location | Substrate | Enzyme | Product |
|---|---|---|---|---|
| 1 | Mitochondria | Succinyl-CoA + Glycine | δ-ALA synthase (requires PLP/B6) | δ-Aminolevulinic acid (δ-ALA) |
| 2 | Cytoplasm | 2× δ-ALA | δ-ALA dehydratase (zinc-dependent) | Porphobilinogen (PBG) |
| 3 | Cytoplasm | 4× PBG | Porphobilinogen deaminase (HMB synthase) | Hydroxymethylbilane (HMB) |
| 4 | Cytoplasm | HMB | Uroporphyrinogen III cosynthase | Uroporphyrinogen III |
| 5 | Cytoplasm | Uroporphyrinogen III | Uroporphyrinogen decarboxylase | Coproporphyrinogen III |
| 6 | Mitochondria | Coproporphyrinogen III | Coproporphyrinogen oxidase | Protoporphyrinogen IX |
| 7 | Mitochondria | Protoporphyrinogen IX | Protoporphyrinogen oxidase | Protoporphyrin IX |
| 8 | Mitochondria | Protoporphyrin IX + Fe²⁺ | Ferrochelatase (heme synthase) | Heme |
Key note on PLP: Step 1 is a decarboxylation/condensation of glycine, requiring pyridoxal phosphate (vitamin B6). A B6 deficiency therefore impairs heme synthesis, causing microcytic hypochromic anemia.
Regulation
Heme regulates its own synthesis via a negative feedback loop on δ-ALA synthase (the rate-limiting enzyme), the first step of the pathway:
- Repression - elevated heme represses transcription of δ-ALA synthase (decreases enzyme synthesis)
- Allosteric inhibition - heme directly inhibits δ-ALA synthase activity
This means heme is synthesized when levels fall, and synthesis decreases as levels rise.
Secondary effect: Heme also stimulates globin synthesis by maintaining the ribosomal initiation complex in an active state, thereby coordinating heme and globin production for hemoglobin assembly.
Drug induction: Drugs like phenobarbital induce cytochrome P450 enzymes, consuming heme. This lowers free heme, which induces δ-ALA synthase, increasing heme synthesis. This mechanism explains why certain drugs can precipitate acute porphyria attacks.
Lead Poisoning and Heme Synthesis
Lead (Pb) inhibits two enzymes:
- δ-ALA dehydratase (step 2, zinc-containing)
- Ferrochelatase (step 8)
Result: accumulation of δ-ALA and protoporphyrin IX, decreased heme production, leading to microcytic hypochromic anemia and neurotoxicity (δ-ALA is structurally similar to GABA and disrupts neuronal signaling).
Porphyrias - Enzyme Deficiencies
Each enzyme deficiency (except step 1 gain-of-function) causes a porphyria, with accumulation of the substrate just before the blocked step:
| Enzyme Deficient | Disease | Clinical Features |
|---|---|---|
| δ-ALA dehydratase | ALA dehydratase porphyria | Neurovisceral (very rare, AR) |
| HMB synthase (PBG deaminase) | Acute Intermittent Porphyria (AIP) | Neurovisceral attacks (AD) |
| Uroporphyrinogen III cosynthase | Congenital erythropoietic porphyria (CEP) | Photosensitivity, hemolysis (AR) |
| Uroporphyrinogen decarboxylase | Porphyria cutanea tarda (PCT) | Skin blistering, most common (AD) |
| Coproporphyrinogen oxidase | Hereditary coproporphyria | Neurovisceral + cutaneous (AD) |
| Protoporphyrinogen oxidase | Variegate porphyria | Neurovisceral + cutaneous (AD) |
| Ferrochelatase | Erythropoietic protoporphyria | Non-blistering photosensitivity (AD) |
| ALAS2 gain-of-function | X-linked protoporphyria | Non-blistering photosensitivity |
Heme Degradation (Brief Overview)
At end of RBC lifespan (~120 days), heme is cleaved by heme oxygenase (producing CO and biliverdin), then reduced to bilirubin, transported by albumin to the liver, conjugated with glucuronic acid, and excreted in bile. Iron is recycled back to stores.
Clinical Mnemonics
- "Succinyl CoA + Glycine → ALA": the committed first step, needs B6
- Lead hits A and F: ALA dehydratase and Ferrochelatase - both inhibited by Pb
- AIP triad: abdominal pain, autonomic dysfunction, axonal neuropathy ("3 A's")
- PCT: most common porphyria; skin fragility and blistering on sun-exposed areas
Sources: Basic Medical Biochemistry - A Clinical Approach 6e, p. 1520-1529; Fitzpatrick's Dermatology Vol. 1, p. 2264; Henry's Clinical Diagnosis and Management by Laboratory Methods.
Porphyria
Finding Sources
Searching the Web
porphyria classification clinical features diagnosis treatment 2024
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Loading Image
Excellent - I now have comprehensive content from Harrison's 22E, Andrews', Fitzpatrick's, and Basic Medical Biochemistry. I have all I need for a thorough answer, including the full pathway diagram. Let me compose the complete response.
Porphyrias
Overview
Porphyrias are a group of largely inherited disorders caused by defects in the heme biosynthetic pathway. Each porphyria results from a partial deficiency of a specific enzyme, causing accumulation of toxic pathway intermediates (porphyrins or their precursors) in tissue, plasma, urine, and/or stool. The specific intermediate that accumulates determines the clinical syndrome.
Classification
Porphyrias are classified by two parallel schemes:
By Site of Accumulation
| Class | Primary Tissue | Examples |
|---|---|---|
| Hepatic | Liver | AIP, HCP, VP, PCT, ADP |
| Erythropoietic | Bone marrow | CEP, EPP, XLP |
By Clinical Presentation
| Class | Presentation | Types |
|---|---|---|
| Acute | Neurovisceral attacks | AIP, HCP, VP, ADP |
| Cutaneous | Photosensitivity / blistering | PCT, CEP, EPP, XLP |
| Mixed (both) | Neurovisceral + cutaneous | HCP, VP |
The Eight Porphyrias - Enzyme, Genetics, Clinic
| Porphyria | Enzyme Deficiency | Inheritance | Key Features |
|---|---|---|---|
| ADP (ALA dehydratase porphyria) | ALA dehydratase | AR | Very rare; neurovisceral |
| AIP (Acute intermittent porphyria) | HMB synthase (PBG deaminase) | AD | Neurovisceral only; no skin |
| CEP (Congenital erythropoietic) | Uroporphyrinogen III synthase | AR | Severe photosensitivity from birth |
| PCT (Porphyria cutanea tarda) | Uroporphyrinogen decarboxylase | AD (type 2) or acquired | Most common porphyria; blistering |
| HCP (Hereditary coproporphyria) | Coproporphyrinogen oxidase | AD | Neurovisceral + cutaneous |
| VP (Variegate porphyria) | Protoporphyrinogen oxidase | AD | Neurovisceral + cutaneous; "South African" |
| EPP (Erythropoietic protoporphyria) | Ferrochelatase | AD | Non-blistering photosensitivity |
| XLP (X-linked protoporphyria) | ALAS2 (gain-of-function) | X-linked | Non-blistering photosensitivity |
Acute Porphyrias - Detailed
Acute Intermittent Porphyria (AIP)
The second most common overall (after PCT) and the most common acute porphyria. Particularly prevalent in Scandinavia. Women outnumber men 1.5-2:1.
Pathophysiology: Deficiency of PBG deaminase (HMB synthase) - 50% activity in affected persons. Only ~10% of gene carriers ever develop clinical disease. δ-ALA and PBG accumulate; these are not photosensitizers (no skin lesions in AIP).
Clinical Features (the "4 A's" triad plus psychiatric):
- Abdominal pain - severe colic, most common initial symptom (up to 95%); no peritoneal signs but tenderness and distention present
- Autonomic dysfunction - tachycardia, hypertension, pupillary dilation, constipation/diarrhea, urinary retention
- Axonal neuropathy - predominantly motor; can progress to flaccid quadriplegia, respiratory paralysis, facial palsy, dysphagia
- Psychiatric - agitation, hallucinations, depression, seizures (10-20%)
Note: Weakness may mimic Guillain-Barré syndrome. EMG shows reduced CMAP amplitudes and active axonal degeneration.
Triggers (all increase hepatic heme demand → induce ALAS1):
- Drugs: anticonvulsants (especially phenytoin, carbamazepine), griseofulvin, rifampin, sulfonamides, barbiturates, progesterone-containing agents
- Hormonal: menstrual cycle (luteal phase), pregnancy
- Dietary: fasting / crash dieting
- Other: infections, surgery, smoking
Diagnosis:
- Acute attack: elevated urinary PBG and δ-ALA (urine may appear red/brown)
- Between attacks: urinary PBG elevated in 88% of cases
- Erythrocyte PBG deaminase activity decreased
- Fecal and erythrocyte porphyrins normal (distinguishes from VP and HCP)
Treatment:
- Remove triggers - stop offending drugs immediately
- Carbohydrate loading - IV glucose ≥300 g/day (downregulates ALAS1); for mild attacks
- IV hemin (heme arginate / Panhematin) - 3-4 mg/kg/day × 4 days; first-line for moderate-severe attacks; suppresses ALAS1 by replacing heme
- Givosiran (Givlaari) - siRNA targeting hepatic ALAS1 mRNA; 2.5 mg/kg SC monthly; FDA/EMA approved 2019 for recurrent acute hepatic porphyrias
- Symptomatic: phenothiazines (nausea/pain), opiates (analgesia), benzodiazepines (seizures - safe in low doses; avoid barbiturates/phenytoin)
- GnRH analogue for luteal-phase attacks
- Liver transplant - last resort for intractable, disabling attacks
Long-term complications: Chronic kidney disease (up to 59% of symptomatic patients), hypertension, hepatocellular carcinoma (HCC) - annual ultrasound + AFP after age 50.
Hereditary Coproporphyria (HCP)
- Enzyme: coproporphyrinogen oxidase deficiency (AD)
- Presents with neurovisceral attacks plus cutaneous blistering (in ~30% of cases)
- Fecal coproporphyrins and protoporphyrins always elevated - key diagnostic marker
- Treatment: same as AIP (hemin, glucose, givosiran); GnRH analogue for premenstrual attacks
Variegate Porphyria (VP) - "Mixed Porphyria"
- Enzyme: protoporphyrinogen oxidase deficiency (AD)
- High prevalence in South Africa (founder effect)
- 40-70% have skin symptoms; 27% have acute attacks; 14% have both
- Skin: blistering vesicles/bullae, hypertrichosis, hyperpigmentation, scarring on sun-exposed areas - resembles PCT
- Neurologic: identical to AIP during attacks
- Pathognomonic: plasma fluorescence peak at 626 nm
- Fecal coproporphyrins + protoporphyrins always elevated; urinary coproporphyrins > uroporphyrins (distinguishes from PCT)
- Increased risk of HCC; liver imaging after age 50
- Treatment: hemin and glucose for acute attacks; antimalarials and phlebotomy not effective for skin in VP
Cutaneous Porphyrias - Detailed
Porphyria Cutanea Tarda (PCT) - Most Common Porphyria
Pathophysiology: Inhibition of hepatic uroporphyrinogen decarboxylase (UROD). Only ~20% of cases have a heterozygous UROD mutation (type 2/familial); the majority are acquired (type 1/sporadic). An inhibitor generated in the liver by iron-catalyzed oxidation inactivates UROD.
Susceptibility factors (acquired):
- Excess iron / HFE hemochromatosis gene mutations
- Alcohol
- Smoking
- Chronic hepatitis C (very strongly associated)
- HIV
- Estrogens (oral contraceptives, HRT)
Clinical features:
- Skin fragility and blistering on sun-exposed areas (dorsa of hands most common)
- Milia, erosions, scarring
- Hypertrichosis (especially facial)
- Hyperpigmentation
Diagnosis:
- Urine: massively elevated uroporphyrins (8-carboxyl porphyrins) - "port wine" urine
- Plasma: elevated highly carboxylated porphyrins
- Stool: elevated isocoproporphyrins (pathognomonic)
- Erythrocyte porphyrins normal (distinguishes from EPP)
Treatment:
- Remove susceptibility factors - abstain from alcohol, estrogens, smoking
- Phlebotomy (300-450 mL every 2-4 weeks) - depletes iron; most reliable therapy; follow serum ferritin to target 15-20 ng/mL
- Low-dose hydroxychloroquine (100-200 mg twice weekly) - chelates porphyrins and promotes urinary excretion; used when phlebotomy is contraindicated (e.g., anemia)
- Treat underlying HCV or HIV
Congenital Erythropoietic Porphyria (CEP) - Günther Disease
- Enzyme: uroporphyrinogen III cosynthase (AR - severe deficiency)
- Presents from birth/infancy with:
- Dark urine (red-pink)
- Severe photosensitivity → vesicles, scarring, mutilation
- Red fluorescence of teeth under Wood's lamp (erythrodontia)
- Hypertrichosis
- Hemolytic anemia
- Treatment: sun protection, blood transfusions; bone marrow transplantation is curative
Hepatoerythropoietic Porphyria (HEP)
- Homozygous form of PCT (homozygous UROD deficiency, ~10% of normal activity)
- Clinical features similar to CEP but biochemistry like PCT
- Bone marrow transplantation may be required
Erythropoietic Protoporphyria (EPP)
- Enzyme: ferrochelatase deficiency (AD with low penetrance; requires second low-expression allele)
- Non-blistering photosensitivity - burning, stinging pain within minutes of sun exposure; edema and erythema
- Liver disease in ~5% (protoporphyrin deposits in bile ducts)
- Diagnosis: elevated free protoporphyrin in erythrocytes; fecal protoporphyrin elevated
- Treatment: sun protection, beta-carotene (some benefit), afamelanotide (MC1R agonist, increases skin melanin)
X-Linked Protoporphyria (XLP)
- ALAS2 gain-of-function mutation → overproduction of protoporphyrin
- Clinically identical to EPP
- X-linked; predominantly males
Biochemical Diagnostic Summary
| Porphyria | Urine ALA | Urine PBG | Urine Porphyrins | Fecal | Erythrocytes |
|---|---|---|---|---|---|
| ADP | ↑↑ | Normal | ↑ Coproporphyrin III | Normal | ↑ Zinc protoporphyrin |
| AIP | ↑↑ | ↑↑↑ | ↑ Uroporphyrin | Normal | ↓ PBGD activity |
| CEP | Normal | Normal | ↑↑ Uroporphyrin I | ↑ Coproporphyrin I | ↑↑ Uroporphyrin |
| PCT | Normal | Normal | ↑↑ Uroporphyrin | ↑ Isocoproporphyrin | Normal |
| HCP | ↑ (attack) | ↑ (attack) | ↑ Coproporphyrin | ↑↑ Coproporphyrin | Normal |
| VP | ↑ (attack) | ↑ (attack) | ↑ Coproporphyrin | ↑↑ Proto + Copro | Normal; plasma 626 nm peak |
| EPP | Normal | Normal | Normal | ↑ Protoporphyrin | ↑↑ Free protoporphyrin |
Safe vs. Unsafe Drugs in Acute Porphyrias
| Category | Safe | Unsafe (Precipitate Attacks) |
|---|---|---|
| Analgesics | Opiates, aspirin, paracetamol | NSAIDs (variable) |
| Sedatives | Benzodiazepines (low dose), chloral hydrate | Barbiturates, chlordiazepoxide |
| Anticonvulsants | Gabapentin, levetiracetam | Phenytoin, carbamazepine, valproate |
| Antibiotics | Penicillins, cephalosporins | Sulfonamides, rifampin |
| Antifungals | Amphotericin B | Griseofulvin |
| Hormones | GnRH analogues | Progesterone, synthetic steroids |
Key Clinical Pearls
- AIP: neurovisceral only, no skin, PBG massively elevated in urine during attacks
- PCT: most common, skin only, blistering on sun-exposed areas, responds to phlebotomy
- VP: "South African porphyria," combination of AIP + PCT features, plasma 626 nm fluorescence is pathognomonic
- EPP/XLP: non-blistering painful photosensitivity, elevated erythrocyte free protoporphyrin
- CEP: rare, most severe, erythrodontia, bone marrow transplant is curative
- Givosiran (2019): first siRNA therapy approved for recurrent acute hepatic porphyria
- All acute porphyria patients need annual liver surveillance after age 50 for HCC
Sources: Harrison's Principles of Internal Medicine 22E (2025), p. 3646; Andrews' Diseases of the Skin 5e, p. 607; Fitzpatrick's Dermatology Vol. 1, p. 2264-2266; Basic Medical Biochemistry - A Clinical Approach 6e, p. 1521-1527
This is a shared conversation. Sign in to Orris to start your own chat.