basics of pituitary gland and Anterior pituitary disorder
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Loading Image
I now have comprehensive material from multiple authoritative textbooks with excellent images. Let me compile the complete answer.
Pituitary Gland - Basics and Anterior Pituitary Disorders
1. ANATOMY AND STRUCTURE
The pituitary gland (hypophysis) is a small, bean-shaped structure weighing approximately 0.5 g (dimensions ~10 × 13 × 6 mm). It sits at the base of the brain within the sella turcica (Turkish saddle) of the sphenoid bone, connected to the hypothalamus by a stalk containing axons and a rich venous (portal) plexus.
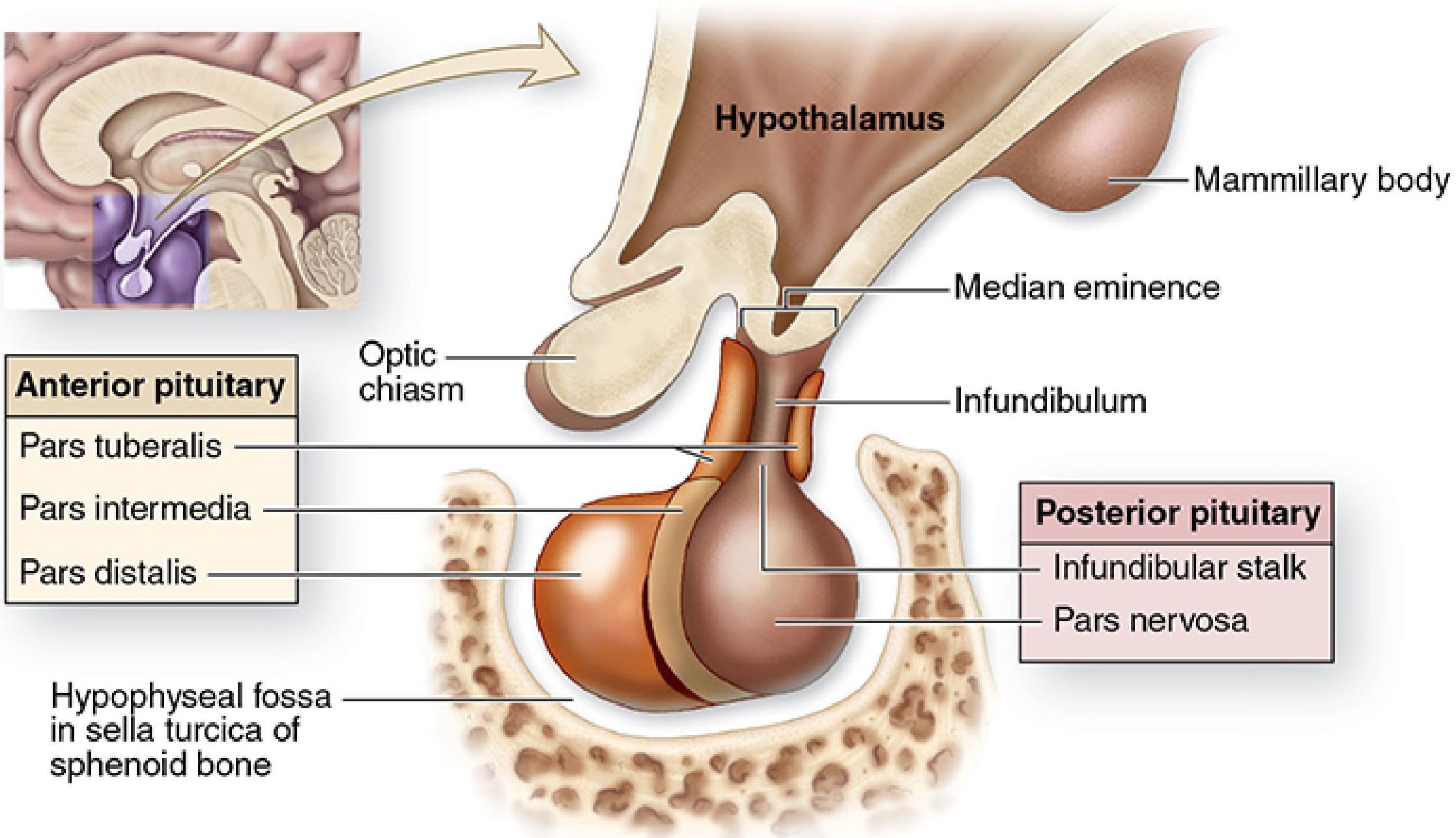
Anatomy of the pituitary gland - Junqueira's Basic Histology, 17e
The gland has two morphologically and functionally distinct parts:
| Feature | Anterior Pituitary (Adenohypophysis) | Posterior Pituitary (Neurohypophysis) |
|---|---|---|
| % of gland | ~80% | ~20% |
| Embryological origin | Rathke's pouch (oral ectoderm) | Neural tissue (downgrowth from diencephalon) |
| Parts | Pars distalis, pars tuberalis, pars intermedia | Pars nervosa + infundibular stalk |
| Cell type | Epithelial secretory cells | Pituicytes (modified glia) + axon terminals |
| Hormones produced here | GH, TSH, ACTH, FSH, LH, PRL | ADH (vasopressin), Oxytocin |
| Note | Hormones synthesized locally | Hormones synthesized in hypothalamus, stored/released here |
2. EMBRYOLOGICAL DEVELOPMENT
During the third week of embryonic development:
- A hypophyseal (Rathke) pouch grows upward from the roof of the pharynx (future anterior pituitary)
- A neurohypophyseal bud grows downward from the diencephalon floor (future posterior pituitary)
By late in the second month, the Rathke pouch detaches from the pharynx and fuses with the neurohypophyseal bud to form the complete gland.
Key transcription factors: POU1F1 (PIT-1) gives rise to somatotrophs, mammosomatotrophs, lactotrophs, and thyrotrophs from a common precursor. Corticotrophs arise from a separate precursor.
3. HISTOLOGY OF THE ANTERIOR PITUITARY
In routine H&E sections, three cell populations are identified based on staining:
- Acidophils (eosinophilic): Somatotrophs (GH) and Lactotrophs (PRL)
- Basophils (basophilic): Corticotrophs (ACTH), Thyrotrophs (TSH), Gonadotrophs (FSH/LH)
- Chromophobes: Poorly staining; may be resting acidophils/basophils or non-secretory cells
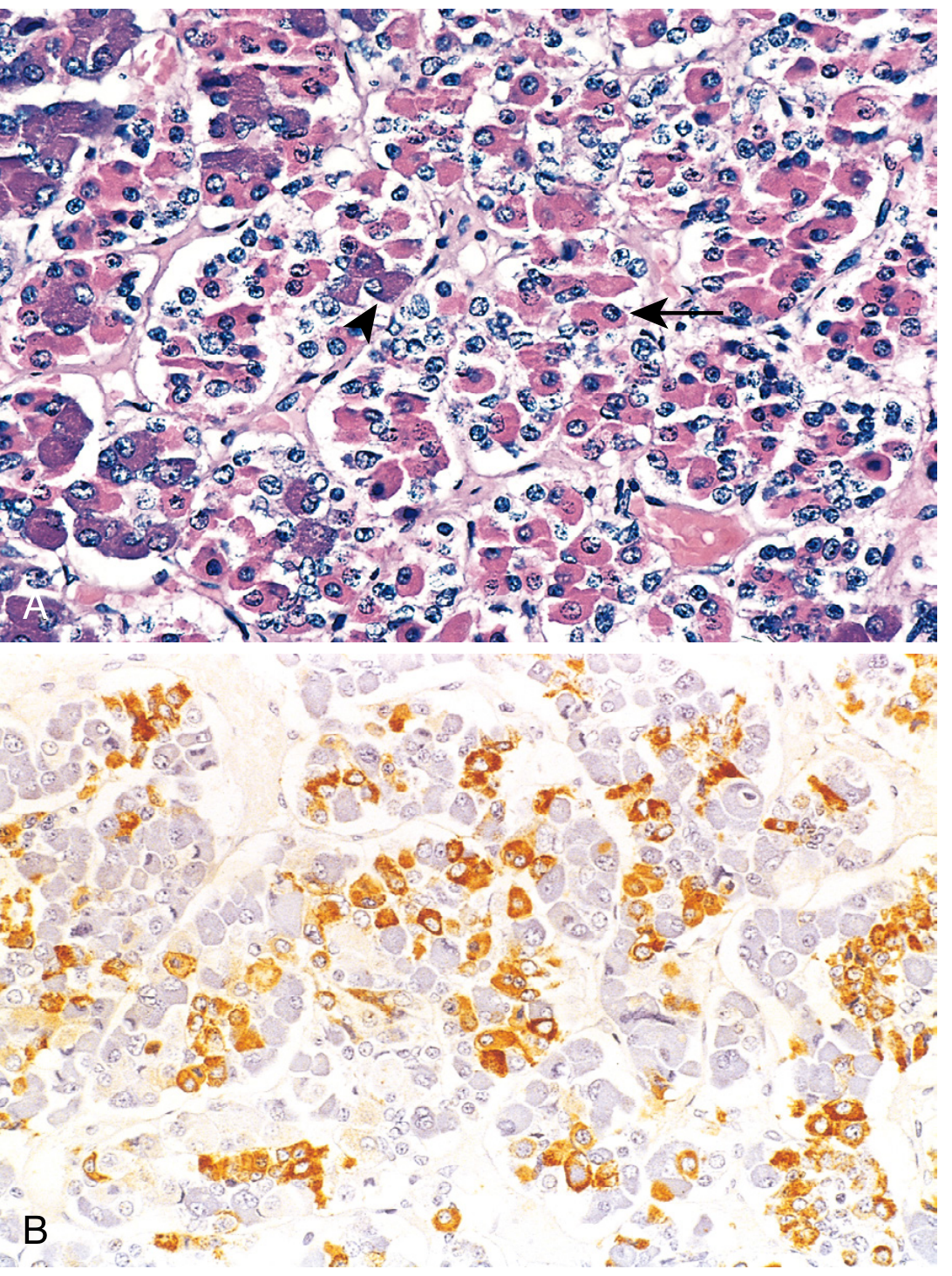
Robbins & Cotran Pathologic Basis of Disease
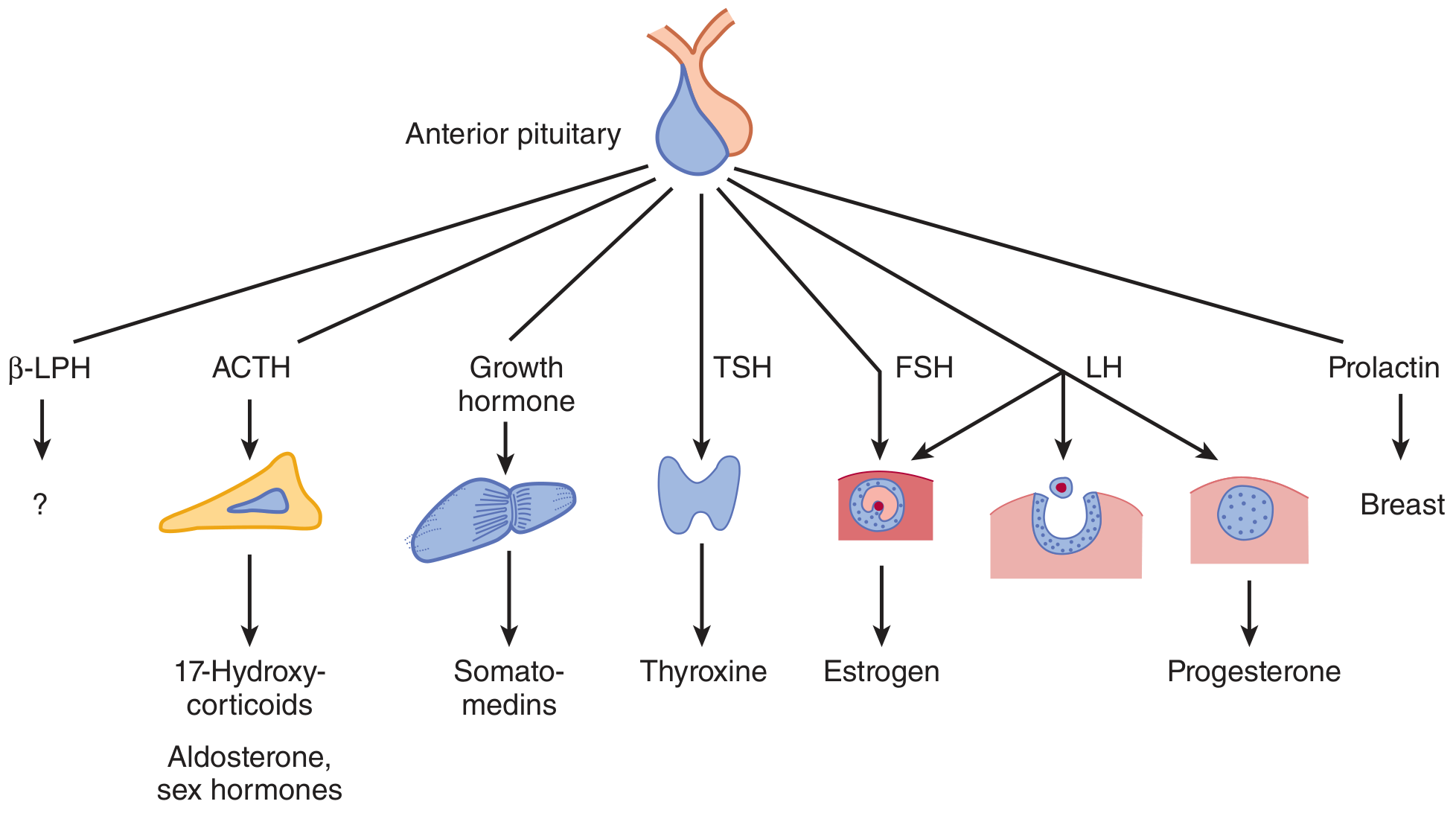
Six Terminally Differentiated Cell Types:
| Cell Type | Hormone | Target |
|---|---|---|
| Somatotrophs | GH (Growth Hormone) | Multiple tissues/liver (IGF-1) |
| Mammosomatotrophs | GH + PRL | |
| Lactotrophs | PRL (Prolactin) | Mammary gland |
| Corticotrophs | ACTH | Adrenal cortex |
| Thyrotrophs | TSH | Thyroid |
| Gonadotrophs | FSH + LH | Ovary, testis |
4. HYPOTHALAMIC-PITUITARY AXIS (Control of Anterior Pituitary)
Anterior pituitary secretion is controlled by hypophysiotropic hormones (releasing/inhibiting factors) produced in the hypothalamus and carried via the portal hypophyseal venous plexus to the anterior pituitary cells.
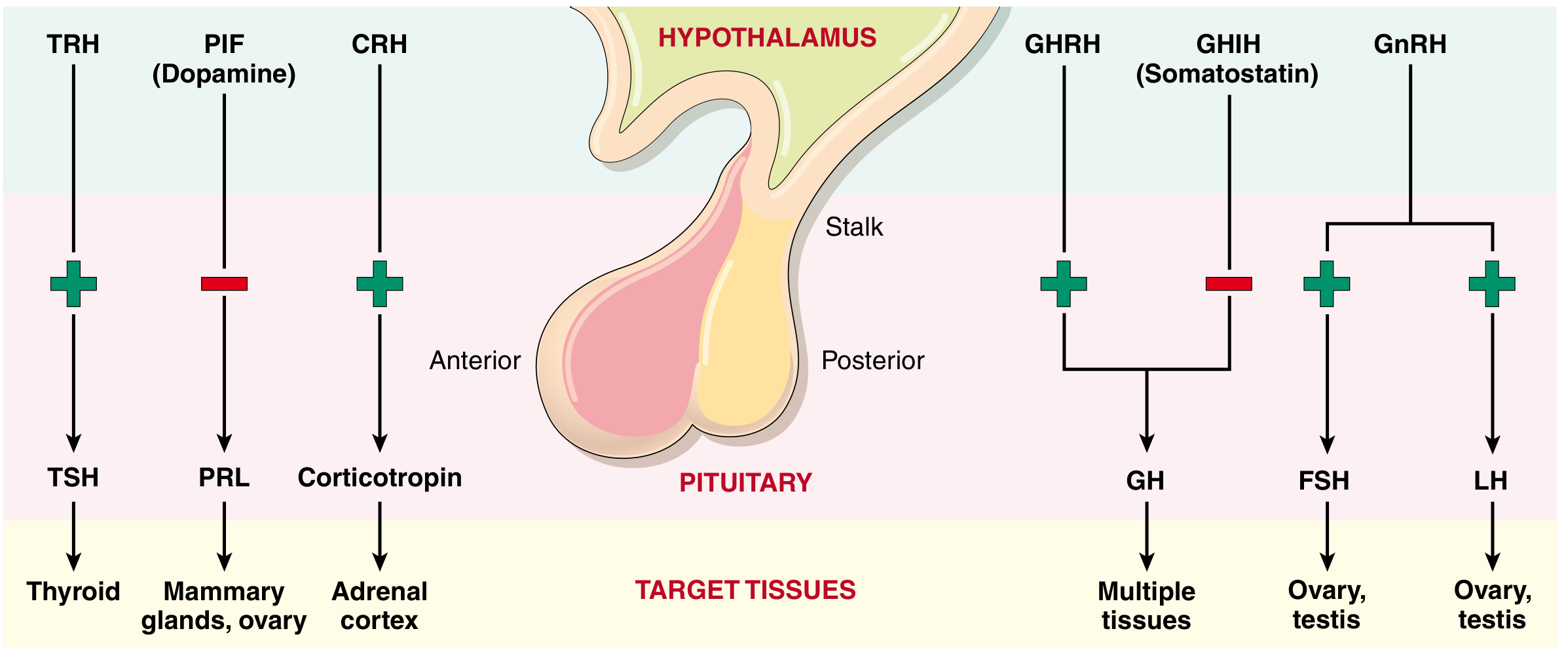
Robbins & Cotran Pathologic Basis of Disease
Hypothalamic Releasing/Inhibiting Hormones:
| Hypothalamic Hormone | Effect | Pituitary Hormone |
|---|---|---|
| CRH (Corticotropin-releasing hormone) | Stimulates (+) | ACTH |
| TRH (Thyrotropin-releasing hormone) | Stimulates (+) | TSH |
| GHRH (GH-releasing hormone) | Stimulates (+) | GH |
| Somatostatin (GHIH) | Inhibits (-) | GH |
| GnRH (Gonadotropin-releasing hormone) | Stimulates (+) | FSH, LH |
| Dopamine (PIF - Prolactin Inhibiting Factor) | Inhibits (-) | PRL |
Ganong's Review of Medical Physiology, 26e
5. CLINICAL MANIFESTATIONS OF PITUITARY DISEASE
Pituitary disorders present through three main mechanisms:
- Hyperpituitarism - excess secretion of trophic hormones (usually from hormone-secreting tumors)
- Hypopituitarism - deficiency of trophic hormones (from ischemia, surgery, radiation, inflammation, or mass effects of non-functional tumors)
- Mass effects - due to proximity to optic chiasm and cranial structures, causing:
- Bitemporal hemianopsia (compression of crossing fibers in the optic chiasm - classic finding)
- Headache, nausea, vomiting (elevated intracranial pressure)
6. ANTERIOR PITUITARY DISORDERS
A. PITUITARY NEUROENDOCRINE TUMORS (PitNETs) and Hyperpituitarism
The current (2017 WHO) classification replaces the old term "pituitary adenoma" with Pituitary Neuroendocrine Tumor (PitNET). They are classified by cell lineage and the transcription factors they express.
Classification by size:
- Microadenoma: < 1 cm
- Macroadenoma: ≥ 1 cm (more likely to cause mass effects)
1. Lactotroph PitNET (Prolactinoma)
- Most common functioning pituitary tumor
- Presents as amenorrhea-galactorrhea in women; impotence and infertility in men
- Serum PRL typically > 200 ng/mL (levels > 100 ng/mL are suggestive)
- Histology: sparse-granule or dense-granule lactotrophs
- Treatment: Dopamine agonists (cabergoline, bromocriptine) - first line; surgery if refractory
2. Somatotroph PitNET (GH-secreting tumor)
- Second most common functioning PitNET
- If excess GH occurs before epiphyseal closure: Gigantism
- If after epiphyseal closure: Acromegaly
- Features: enlarged hands/feet, coarse facial features, prognathism, macroglossia, increased sweating, hypertension, diabetes mellitus, carpal tunnel syndrome, sleep apnea
- Diagnosis: elevated IGF-1 levels; GH not suppressed with oral glucose tolerance test
- Genetics: GNAS somatic activating mutation (most common genetic alteration)
- Treatment: surgery (transsphenoidal resection); somatostatin analogs (octreotide, lanreotide); pegvisomant (GH receptor antagonist)
3. Corticotroph PitNET (ACTH-secreting tumor)
- Causes Cushing disease (hypercortisolism of pituitary origin - one cause of Cushing syndrome)
- Features: truncal obesity, moon facies, buffalo hump, abdominal striae, hypertension, osteoporosis, glucose intolerance, hypokalemia, psychiatric changes
- Usually microadenomas (< 1 cm) - hard to detect on MRI
- Genetics: USP8 somatic activating mutation (most common in corticotroph tumors)
- Diagnosis: elevated 24-hour urinary free cortisol; elevated midnight salivary cortisol; failure of low-dose dexamethasone suppression test; elevated ACTH
- Note: Nelson syndrome - after bilateral adrenalectomy for Cushing disease, removal of adrenal feedback leads to rapid growth of the pre-existing corticotroph tumor with skin hyperpigmentation
4. Thyrotroph PitNET (TSH-secreting tumor)
- Rarest functioning PitNET
- Causes central (secondary) hyperthyroidism
- Features: hyperthyroidism with elevated TSH (paradoxical - TSH should be suppressed in primary hyperthyroidism)
5. Gonadotroph PitNET (FSH/LH-secreting tumor)
- Usually non-functional (silent) - presents with mass effects and hypopituitarism
- Rarely causes hormonal syndrome
- Most common non-functioning macroadenoma subtype
6. Null Cell Tumor
- No identifiable hormone expression on immunostaining
- Present only with mass effects (visual field defects, headache, hypopituitarism)
B. HYPOPITUITARISM
Deficiency of one or more anterior pituitary hormones.
Causes:
- Sheehan syndrome - postpartum pituitary necrosis due to ischemia following massive hemorrhage (most common cause of hypopituitarism in women)
- Mass effects from non-functional tumors compressing normal parenchyma
- Surgery or radiation to pituitary region
- Inflammatory/infiltrative disease (sarcoidosis, lymphocytic hypophysitis, hemochromatosis)
- Pituitary apoplexy - hemorrhage or infarction into a pituitary tumor (acute emergency)
- Empty sella syndrome
Clinical Features (ordered by hormone sensitivity to destruction - GH lost first, ACTH last):
| Hormone Lost | Clinical Effect |
|---|---|
| GH | Growth failure in children; fatigue, reduced muscle mass in adults |
| FSH/LH | Hypogonadism, infertility, amenorrhea, loss of libido |
| TSH | Secondary hypothyroidism (fatigue, cold intolerance, weight gain) |
| ACTH | Secondary adrenal insufficiency (weakness, weight loss, hypotension, hypoglycemia) - life-threatening |
| PRL | Inability to lactate postpartum |
Note: In secondary adrenal insufficiency (ACTH deficiency), there is NO hyperpigmentation (unlike primary Addison disease), because ACTH/MSH is LOW.
C. GENETIC SYNDROMES ASSOCIATED WITH PITUITARY TUMORS
| Gene | Syndrome | Associated Tumor |
|---|---|---|
| MEN1 | Multiple Endocrine Neoplasia type 1 | Somatotroph, lactotroph, or corticotroph |
| PRKAR1A | Carney complex | Somatotroph or lactotroph |
| AIP | Familial isolated pituitary NET (FIPA) | Somatotroph or lactotroph (especially < 35 years) |
| CDKN1B (p27/KIP1) | "MEN1-like" syndrome | Corticotroph |
| DICER1 | DICER1 syndrome | Pituitary blastoma |
7. SUMMARY TABLE - Anterior Pituitary Hormones and Disorders
| Hormone | Excess | Deficiency |
|---|---|---|
| GH | Gigantism (child), Acromegaly (adult) | Growth failure (child), fatigue/body composition changes (adult) |
| PRL | Amenorrhea-galactorrhea, infertility, impotence | Failure of postpartum lactation |
| ACTH | Cushing disease | Secondary adrenal insufficiency |
| TSH | Central hyperthyroidism | Secondary hypothyroidism |
| FSH/LH | Rarely symptomatic | Hypogonadism, infertility |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Ganong's Review of Medical Physiology, 26e; Junqueira's Basic Histology, 17e; Guyton and Hall Textbook of Medical Physiology
This is a shared conversation. Sign in to Orris to start your own chat.