Teache me as neonatatologist, paeditrcian in depth topic- Meningocele
Reading File
Finding Sources
Finding Sources
Searching PubMed
"meningocele" OR "myelomeningocele" AND "neonatal management"
Reading File
Reading File
Finding Sources
Finding Sources
Searching PubMed
"spina bifida" AND "neonatal" AND management
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
I now have comprehensive material from multiple authoritative textbooks. Let me compile the full in-depth teaching response for a neonatologist/paediatrician.
Meningocele - In-Depth Teaching for Neonatologists & Paediatricians
1. DEFINITION & CLASSIFICATION - Spectrum of Neural Tube Defects (NTDs)
Meningocele sits within the broader spectrum of spinal dysraphism - disorders of failed midline fusion of dorsal neural tube structures, which takes place during the first 3 weeks of postconceptual life.
| Condition | Contents of Sac | Neural Tissue in Sac? | Neurological Status |
|---|---|---|---|
| Spina bifida occulta | None - no external sac | No | Usually normal |
| Meningocele | Dura + arachnoid + CSF | No - cord remains in canal | Usually neurologically NORMAL |
| Myelomeningocele (MMC) | Dura + arachnoid + CSF + cord/cauda equina | YES | Significant neurological deficits |
| Encephalocele | Cranial - meninges ± brain tissue | Sometimes | Variable |
Key distinction for neonatologists: In meningocele, there is a protrusion of only the dura and arachnoid through the defect in the vertebral laminae, forming a cystic swelling - the cord remains in the canal. In myelomeningocele, which is 10 times more frequent than meningocele, the cord (more often the cauda equina) is extruded and closely applied to the fundus of the cystic swelling. Most patients with meningocele are neurologically normal, unlike MMC.
- Adams and Victor's Principles of Neurology, 12th Ed.; Schwartz's Principles of Surgery 11th Ed.
2. EMBRYOLOGY & PATHOPHYSIOLOGY
Normal Neural Tube Closure
- Neural tube closes between days 22-28 post-fertilisation (postconceptual days 3-4 weeks)
- Closure proceeds in a zipper-like fashion from the cervical region cranially and caudally
- Failure of closure at various levels produces different defects
The "Two-Hit Hypothesis" (for myelomeningocele, but conceptually relevant)
- First hit: Neural tube fails to close → CSF leakage + neural placode exposure
- Second hit: Direct trauma from uterine wall contact + chemical toxicity of amniotic fluid causes acquired injury
Understanding this distinction matters for meningocele too: since no neural tissue is extruded, meningocele largely avoids the second-hit neurological injury - explaining why most meningocele patients are neurologically intact.
- Mulholland & Greenfield's Surgery 7e
Sites
- Most common: lumbosacral region
- Can also occur: cervical, thoracic (lateral thoracic meningocele), sacral (anterior sacral meningocele), cranial (cranial meningocele/encephalocele)
3. EPIDEMIOLOGY & INCIDENCE
- Spinal dysraphism incidence varies widely by geography/locale
- Risk of recurrence: if one child affected → rises from ~1/1,000 to 40-50/1,000 (a 30-fold increase) for neural tube defects
- Myelomeningocele alone: ~4 children born per day in the United States
- Meningocele is significantly less common than MMC (ratio ~1:10)
- Sex: slightly more common in females
- More common in populations with lower folate intake
Risk Factors (from urology/Campbell-Walsh and Adams & Victor)
- Maternal folate deficiency (most important modifiable risk)
- Antiepileptic drugs: valproic acid and carbamazepine (folic acid antagonists)
- Maternal diabetes and obesity
- Maternal fever/flu during early pregnancy
- Advanced or very young maternal age
- Previous miscarriage, low socioeconomic status
- Maternal passive smoking, caffeine consumption
- Strong familial risk: mother with one affected child has 20-50x increased risk
- Campbell-Walsh-Wein Urology
4. ANTENATAL DETECTION & DIAGNOSIS
Screening Tools
| Test | Details |
|---|---|
| AFP in amniotic fluid | Elevated at 15-16 weeks gestation (open NTDs leak AFP into amniotic fluid) |
| Maternal serum AFP | Elevated in open NTDs |
| Acetylcholinesterase assay | Done on amniotic fluid - reliable confirmation of neural tube defects |
| Fetal ultrasound | Confirms and characterises defect; standard of care from 18-20 weeks anomaly scan |
| MRI (fetal) | Better soft tissue characterization, increasingly used |
Important: Blood contamination is a source of error in the AFP test. Acetylcholinesterase immunoassay is the more reliable confirmatory test.
- Adams and Victor's Principles of Neurology
Ultrasound Signs in NTDs
- Lemon sign: Scalloping of frontal bones (characteristic of open NTDs)
- Banana sign: Curved/effaced cerebellum pulled down into foramen magnum (Chiari II)
- Posterior fossa abnormalities
- Direct visualisation of sac in lumbosacral region
5. CLINICAL PRESENTATION AT BIRTH - NEONATAL ASSESSMENT
Physical Examination
The newborn presents with a midline dorsal cystic swelling, most often in the lumbosacral region:
- Soft, fluctuant, translucent cystic mass in the midline
- Covered by intact skin (more common) OR thinned/weeping skin (higher risk of rupture)
- Transilluminates (critical distinction from lipoma/teratoma which do not)
- May enlarge with crying (increased intracranial pressure transmitted)
- No neural tissue visible or palpable in the base of sac (distinguishes from MMC)
Cutaneous Signs on the Back - Know These!
Per the dermatology atlas (Fitzpatrick's), meningoceles and spinal dysraphism may be preceded or accompanied by midline cutaneous markers:
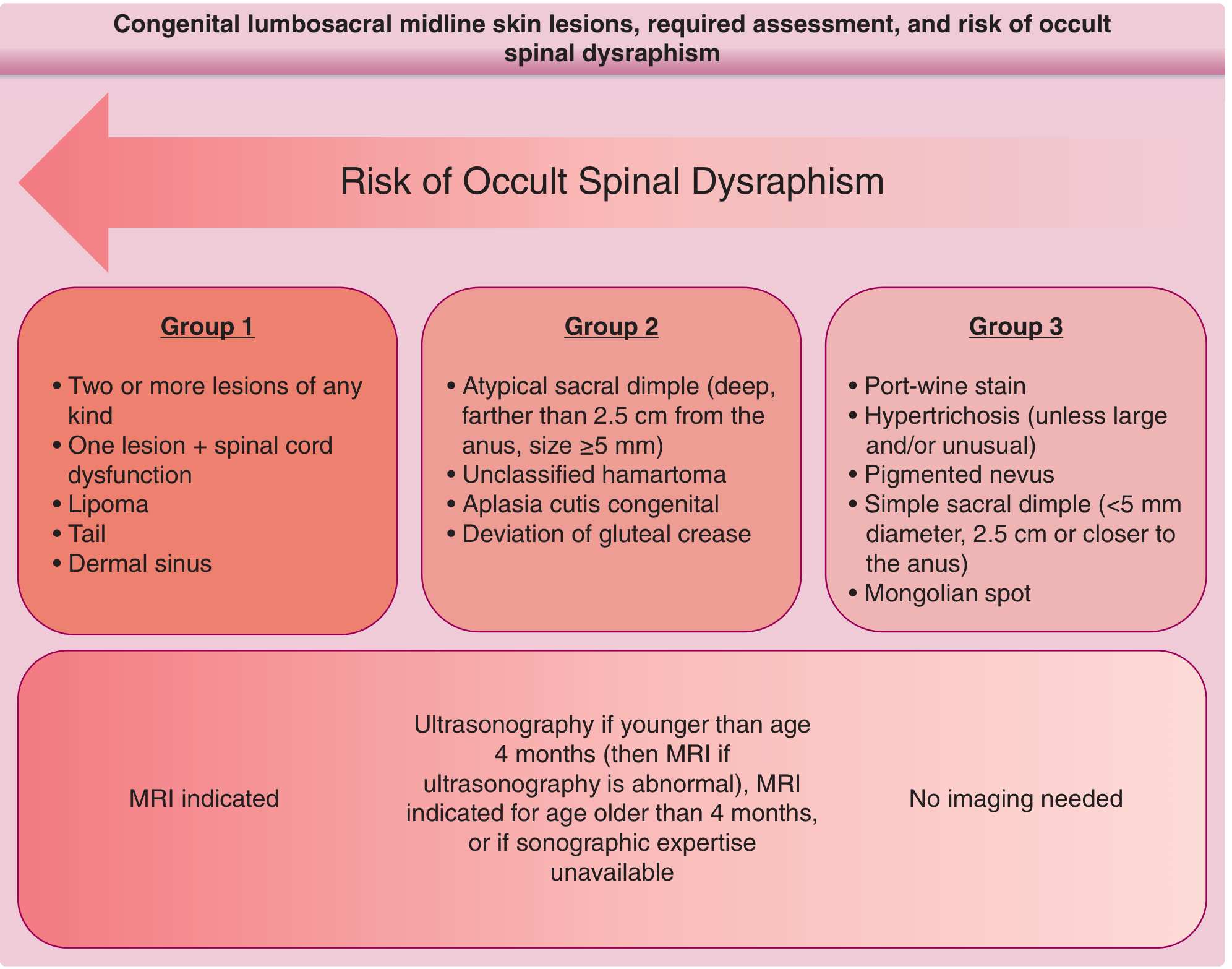
Fitzpatrick's Dermatology - Risk stratification for occult spinal dysraphism based on skin lesion type
High-risk cutaneous markers (Group 1 - MRI indicated):
- Lipoma overlying the spine
- Dermal sinus
- Tail (true human tail)
- Two or more lesions of any type
- Any lesion + spinal cord dysfunction
Intermediate risk (Group 2 - Ultrasound <4 months, then MRI):
- Atypical sacral dimple (>5mm, farther than 2.5cm from anus)
- Aplasia cutis congenita
- Deviation of gluteal crease
- Unclassified hamartoma
Lower risk (Group 3 - No imaging needed):
- Simple sacral dimple (<5mm, within 2.5cm of anus)
- Port-wine stain, pigmented nevus
- Mongolian spot
Neurological Examination
In pure meningocele (no neural tissue herniated):
- Lower limb movements: normal
- Tone: normal
- Reflexes: present and normal
- Bladder/bowel function: typically normal
- Response to plantar stimulus: normal
If neurological deficits ARE found → suspect myelomeningocele, not meningocele → reclassify and escalate urgency.
6. IMAGING IN THE NEONATE
Modality of Choice: MRI
MRI is the gold standard - it precisely:
- Confirms cord position (is it in the canal or herniated into the sac?)
- Identifies the level and extent of vertebral defect
- Detects associated anomalies (Chiari malformation, tethered cord, syrinx)
- Rules out neural tissue in sac (meningocele vs. MMC)
- Shows ventricular size (screening for hydrocephalus)
Pre-operative MRI is mandatory before surgical excision and reconstruction of meningocele, as it may be associated with a persistent intracranial defect. - Fitzpatrick's Dermatology
Other Imaging
- Spinal ultrasound (preferred <4 months as posterior arches not yet ossified - acoustic window available)
- Can assess cord position, pulsations, tethering
- Limitations: operator-dependent, misses subtle tethering
- Plain X-ray spine: Shows the posterior vertebral arch defect (bifid spine)
- CT: Less preferred due to radiation; CT myelography used if MRI contraindicated
- Cranial ultrasound: Screen for hydrocephalus in all NTDs at birth
Anterior Sacral Meningocele (special variant)
- Cystic presacral mass communicating with the subarachnoid space
- Plain X-ray: pathognomonic "scimitar sacrum" - eccentric anterior sacral defect
- MRI best demonstrates communication with sacral canal
- Grainger & Allison's Diagnostic Radiology
Lateral Thoracic Meningocele (special variant)
- Presents as a paravertebral mass on CXR
- 70-85% associated with neurofibromatosis - must screen!
- Angular kyphoscoliosis + pressure erosion of intervertebral foramen
- Grainger & Allison's Diagnostic Radiology
7. ASSOCIATED CONDITIONS - WHAT TO SCREEN FOR
Every neonate with a meningocele or suspected spinal dysraphism needs systematic evaluation:
| System | Association | Why it matters |
|---|---|---|
| CNS - Hydrocephalus | Common with MMC, less so in pure meningocele | VP shunt may be needed |
| CNS - Chiari II malformation | Downward herniation of cerebellum + brainstem through foramen magnum | Risk of obstructive apnoea, swallowing dysfunction |
| Urological - neurogenic bladder | Very common in MMC; check even in meningocele | Silent hydronephrosis, UTIs, renal failure |
| Orthopaedic - hip dislocation | Due to muscle imbalance | Club foot, kyphoscoliosis |
| Cardiac anomalies | ~5-10% associated CHD | Echo at birth |
| Chromosomal | Trisomy 18, 13 rarely | Karyotype if dysmorphic |
| Tethered spinal cord | Even after repair | Delayed neurological deterioration |
Almost all infants born with spina bifida (especially MMC) have an Arnold-Chiari malformation, which includes hindbrain herniation, brainstem abnormalities, low-lying venous sinuses, and a small posterior fossa. - Campbell-Walsh-Wein Urology
8. NEONATAL MANAGEMENT - IMMEDIATE (FIRST 24-48 HOURS)
A - Airway & Breathing
- Assess for apnoea or stridor - may indicate associated Chiari II malformation with brainstem compression
- If present → urgent neurosurgical consultation
B - Protect the Sac
- Position prone or lateral to avoid pressure on sac
- Cover with sterile saline-moistened non-adherent gauze
- Wrap loosely - do not constrict
- Do NOT rupture the sac
C - Infection Prevention
- IV antibiotics (typically ampicillin + gentamicin) to prevent meningitis, especially if sac appears thin or leaking CSF
- The two most common complications of spinal defects are meningitis and progressive hydrocephalus
- Excision and closure in the first few days of life is advised to prevent fatal meningitis
D - Investigations
- Full blood count, CRP, blood culture, LFTs, urea/creatinine
- Cranial ultrasound (hydrocephalus screening)
- Spinal ultrasound or MRI spine
- Renal ultrasound (baseline for neurogenic bladder)
- Urine output monitoring
- Echocardiogram (screen for CHD)
E - Team Assembly - Multidisciplinary
- Neonatologist
- Paediatric neurosurgeon (primary surgical team)
- Paediatric urologist
- Paediatric orthopaedics
- Paediatric physiotherapy
- Social work / parent counselling
9. SURGICAL MANAGEMENT
Timing
- Within 24-72 hours of birth for open/ruptured lesions (meningitis risk)
- For intact-skinned meningocele with no CSF leak: can be done semi-electively within days-weeks, but early repair is still standard practice
Procedure
- MRI first to map anatomy
- Surgical excision of the herniated sac
- Careful preservation of any neural elements adherent to sac (rarely present in pure meningocele)
- Watertight dural closure
- Multilayer skin closure
Outcomes for Meningocele (vs. MMC)
- Most patients with pure meningocele are neurologically normal post-repair
- Re-tethering of cord is a late complication
- Far better than MMC: the Lorber series of MMC showed 80-90% of surviving patients were developmentally delayed and paraplegic
- Exceptionally, patients with meningomyelocele, and most of those with lumbar meningocele, are cognitively normal
- Adams and Victor's Principles of Neurology
Hydrocephalus Management
- VP shunt: traditional standard
- Endoscopic third ventriculostomy (ETV) + choroid plexus cauterization (CPC): effectively manages hydrocephalus in >70% of patients with MMC, avoids VP shunt, with similar neurocognitive outcomes
- Campbell-Walsh-Wein Urology (citing Warf & Campbell, 2008)
Fetal (Prenatal) Surgery - MOMS Trial (for MMC, relevant background)
- The MOMS trial (Management of Myelomeningocele Study) showed:
- 50% decrease in need for VP shunting
- Decreased Chiari neurologic malformations
- 42% of infants walking at 30 months vs. 21% in postnatal repair group
- This is for MMC, not pure meningocele, but represents the frontier - benefits and complications of fetal/postnatal surgery for open spina bifida - a 2025 meta-analysis (PMID 40492626)
- In 2020, first-in-human FDA-approved trial for placental mesenchymal stem cells (PMSCs) seeded on dural graft ECM for fetal MMC repair (CuRE study)
- Mulholland & Greenfield's Surgery 7e
10. LONG-TERM FOLLOW-UP & COMPLICATIONS
Urological (Even in Pure Meningocele - Monitor)
- Serial renal ultrasounds
- Urodynamic studies - assess bladder function
- Clean intermittent catheterisation (CIC) if neurogenic bladder develops
- Prophylactic antibiotics for recurrent UTIs
- Goal: preserve upper tract (kidneys)
Neurological
- Serial developmental assessments
- Monitor for late tethered cord syndrome:
- Progressive scoliosis
- Worsening bladder/bowel function
- Back pain
- New lower limb weakness
- Requires surgical detethering
Orthopaedic
- Hip surveillance
- Foot deformity (clubfoot, cavus)
- Scoliosis screening
Neurodevelopmental
- Cognitive assessment
- Educational support as needed
- In pure meningocele: usually normal cognitive development
11. TYPES OF MENINGOCELE - SUMMARY TABLE
| Type | Location | Key Features | Association |
|---|---|---|---|
| Posterior spinal | Lumbosacral (most common) | Posterior midline cystic mass, transilluminates | Most common form |
| Sacral/presacral | Anterior sacral | "Scimitar sacrum" on X-ray, pelvic mass | Currarino triad |
| Lateral thoracic | Paravertebral | CXR: paravertebral mass, kyphoscoliosis | 70-85% neurofibromatosis |
| Anterior thoracic | Ventral cord herniation | Chronic thoracic myelopathy | Rare |
| Cranial/occipital | Posterior skull defect | Meninges only (no brain tissue) | Can be part of encephalocele spectrum |
| Rudimentary meningocele | Skin only | Small cutaneous lesion, remnant | Occult dysraphism |
12. PREVENTION
Folic Acid Supplementation - The Most Important Intervention
- Folic acid before the 28th day of pregnancy is protective (neural tube closes by day 28)
- Standard recommendation: 0.4-0.8 mg/day periconceptionally for all women of reproductive age
- High-risk women (prior NTD pregnancy, anticonvulsant use): 4-5 mg/day starting at least 1 month before conception
- Folic acid grain fortification programmes have resulted in a 20-50% decrease in NTD prevalence
- Vitamin A may have slight additional protective benefit
Antiepileptic Drug Management
- Switch to safer AEDs in women of childbearing age before pregnancy
- Folic acid supplementation is essential when valproate/carbamazepine cannot be avoided
13. KEY DIFFERENTIALS - NEONATAL POSTERIOR MIDLINE MASS
| Diagnosis | Features |
|---|---|
| Meningocele | Soft, fluctuant, transilluminates, midline lumbosacral, cord in canal, normal neurology |
| Myelomeningocele | Neural tissue in sac, neurological deficits, motor/sensory loss |
| Lipomyelomeningocele | Fatty mass, skin covered, partial tethering |
| Sacrococcygeal teratoma | Solid ± cystic, does not transilluminate uniformly, AFP elevated |
| Pilonidal sinus / dermoid | No transillumination, may have hair, midline pit |
| Haemangioma/vascular malformation | Vascular, may be pulsatile |
| Lipoma | Solid, non-transilluminant, soft |
14. QUICK EXAM PEARLS
- Meningocele = meninges + CSF only (cord in canal) - most are neurologically normal
- Meningomyelocele = 10x more common than meningocele
- Most common site: lumbosacral
- Folic acid must be given before 28 days post-conception to be effective
- Anterior sacral meningocele = pathognomonic "scimitar sacrum" on X-ray
- Lateral thoracic meningocele = 70-85% associated with neurofibromatosis
- Always check for Chiari II, hydrocephalus, neurogenic bladder even if sac looks "simple"
- MRI spine mandatory pre-op
- ETV + CPC can now manage hydrocephalus without VP shunt in >70% of cases
- Arnold-Chiari malformation is present in almost ALL patients with spina bifida
- Valproate and carbamazepine - major teratogenic risk factors for NTDs
Sources:
- Adams and Victor's Principles of Neurology, 12th Ed.
- Mulholland & Greenfield's Surgery, 7e - Myelomeningocele and Neural Tube Defects
- Schwartz's Principles of Surgery, 11th Ed.
- Campbell-Walsh-Wein Urology - Neurogenic Bladder in NTDs
- Fitzpatrick's Dermatology, Vol 1-2 - Cutaneous markers of spinal dysraphism
- Andrews' Diseases of the Skin - Encephalocele and Meningocele
- Grainger & Allison's Diagnostic Radiology - Spinal Meningoceles
- Recent literature: Kunpalin Y, et al., 2025 - Fetal vs. postnatal surgery for open spina bifida (PMID 40492626)
Teache me as neonatatologist, paeditrcian in depth topic- Meningomyelocele
Reading File
Finding Sources
Finding Sources
Searching PubMed
"myelomeningocele" AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive, multi-source content. Let me compile the full in-depth teaching response.
Myelomeningocele (MMC) - In-Depth Teaching for Neonatologists & Paediatricians
1. DEFINITION & OVERVIEW
Myelomeningocele (MMC) is the herniation of the spinal cord AND meninges (dura + arachnoid + CSF) through a congenital posterior vertebral arch defect. It is the most common form of spina bifida and the most significant open neural tube defect (NTD).
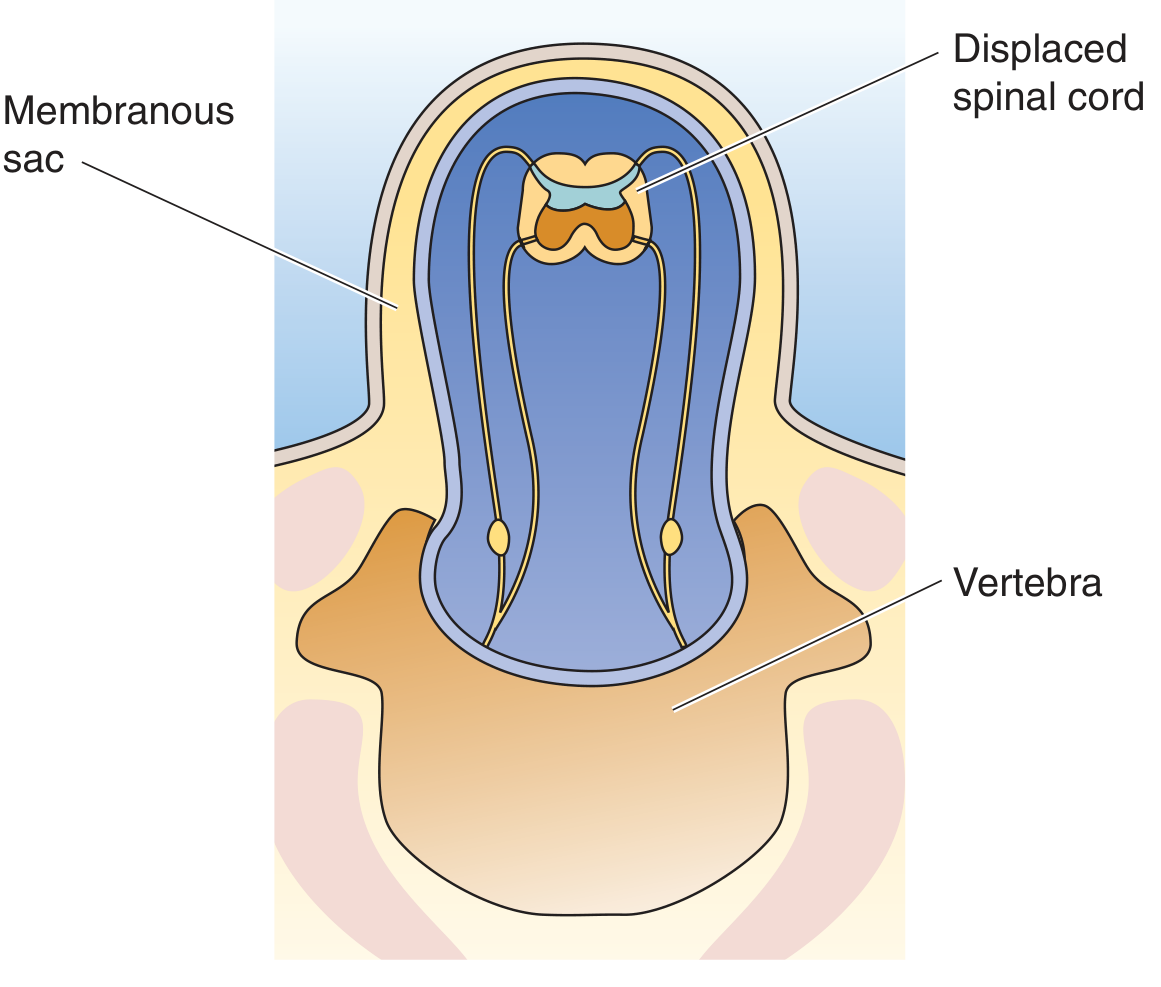
Medical Physiology (Boron & Boulpaep) - Fig. 10-7: Myelomeningocele. The spinal cord AND meninges herniate through the vertebral defect into a membranous sac.
Classification of Neural Tube Defects (Complete Spectrum)
| Type | Contents | Neurological Status |
|---|---|---|
| Spina bifida occulta | No sac; vertebral arch defect only | Normal |
| Meningocele | Dura + arachnoid + CSF only | Usually normal |
| Myelomeningocele | Dura + arachnoid + CSF + spinal cord/nerve roots | Always deficits |
| Lipomyelomeningocele | Fat + cord herniation, skin-covered | Variable |
| Anencephaly | Absent brain + skull | Lethal |
| Encephalocele | Cranial - meninges ± brain tissue | Variable |
Medical Physiology - Table 10-3: Defects of Neural Tube Closure
2. EPIDEMIOLOGY
- Incidence: ~1 in 3,000 live births (USA); ~9:10,000 births worldwide for all NTDs
- In the USA: approximately 4 children born daily with MMC
- One of the most common congenital defects causing lifelong paralysis
- Sex: Slightly more common in females
- Geography: Higher prevalence in Ireland, Wales, India, South America; lower in Africa, Asia
- 5-year mortality rate among those undergoing neonatal repair: 79:1,000
- 25-40% of MMC pregnancies are terminated after prenatal diagnosis
- >80% of children require lifelong VP shunting
- Only 37% of survivors can live independently as adults; >70% have IQ >80
- Creasy & Resnik's Maternal-Fetal Medicine; Miller's Anesthesia 10e
3. EMBRYOLOGY & PATHOPHYSIOLOGY
Normal Neural Tube Closure
- Neural tube forms and closes between days 22-28 post-fertilisation
- Closure progresses bidirectionally from the cervical region
- MMC results from failure of primary neurulation - specifically distal neural tube closure
The Two-Hit Hypothesis
This is the core pathophysiological framework for MMC:
First Hit - Primary Malformation:
- Failure of neural tube closure during the 4th week of gestation
- Neural placode (exposed neural tissue) remains open
- CSF leaks from the spinal defect
- Leads to hindbrain herniation, obstruction of CSF flow, and hydrocephalus
Second Hit - Acquired Injury (in utero):
- Exposed neural placode subjected to:
- Direct mechanical trauma from uterine wall contact
- Neurotoxic damage from amniotic fluid
- This progressive injury worsens neurological function during gestation
- Ultrasound confirms: lower limb movement may be lost and hydrocephalus worsens during pregnancy
- This is the rationale for fetal surgery - preventing the second hit
Mulholland & Greenfield's Surgery 7e; Sabiston Textbook of Surgery; Creasy & Resnik's MFM
Upstream Brain Effects
The abnormal spinal cord anatomy exerts downward displacement on the cerebellar vermis and brainstem, pulling them into the spinal canal. This produces the Chiari II malformation, present in virtually all MMC patients. The Chiari II in turn impairs CSF circulation through the fourth ventricle → secondary obstructive hydrocephalus.
4. ANTENATAL DETECTION & DIAGNOSIS
Prenatal Screening
| Test | Details | Clinical Value |
|---|---|---|
| Maternal serum AFP | Elevated at 15-18 weeks | Screening test |
| Amniotic fluid AFP | Higher sensitivity than maternal serum | Confirmatory |
| Acetylcholinesterase (AChE) immunoassay | Done on amniotic fluid | Most reliable confirmation |
| 2nd trimester ultrasound (18-20 wks) | Standard of care | Morphological diagnosis |
| Fetal MRI | Superior soft tissue detail | Characterise lesion, brain anomalies |
Classic Ultrasound Signs
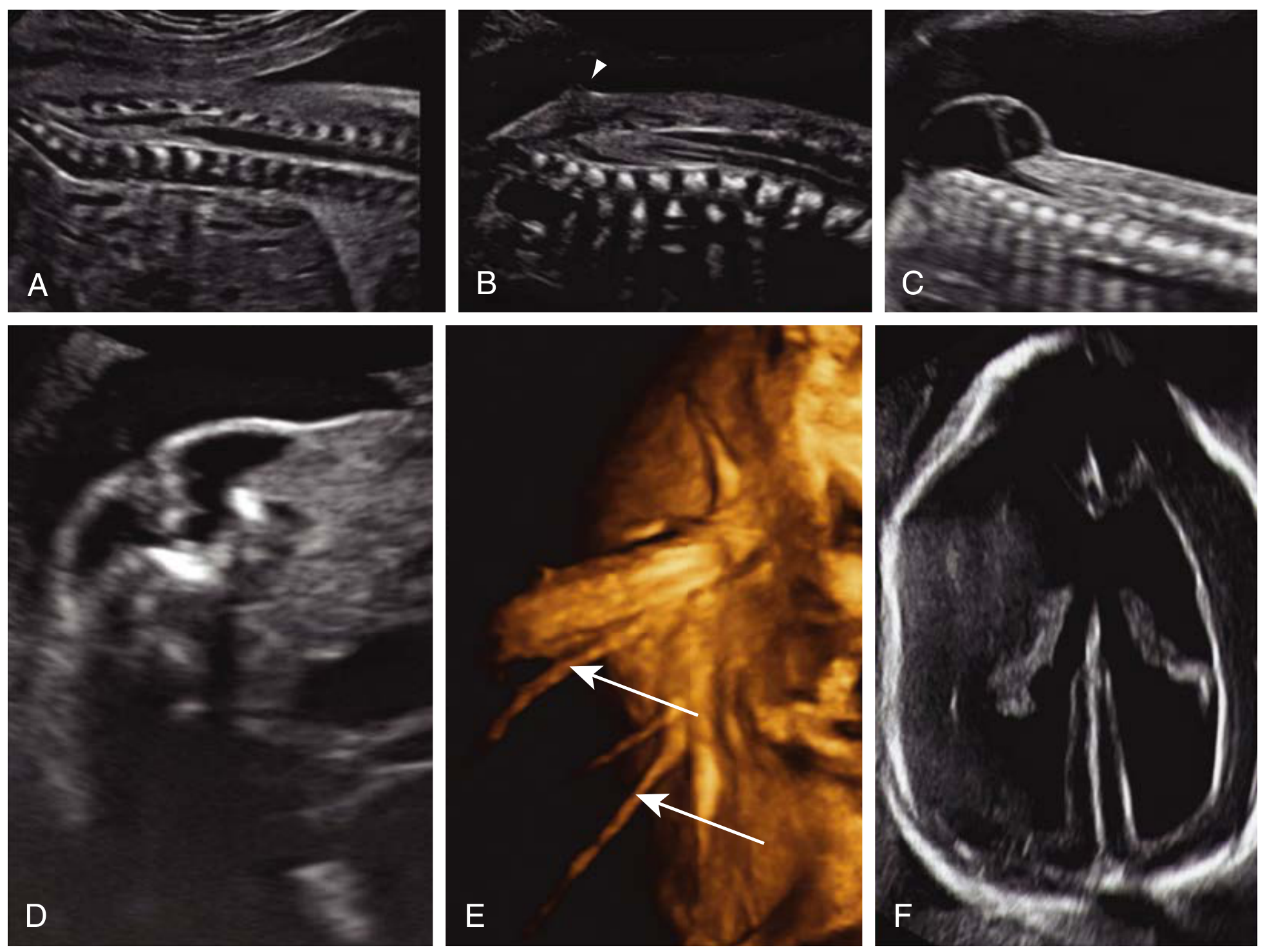
Creasy & Resnik's MFM - Fig. 34.16: Prenatal ultrasound findings in MMC. (A) Normal conus medullaris at L2-L3. (B) Tethered cord/lipoma with conus at L4, dorsal position. (C) Cystic lumbosacral mass on midsagittal section. (D) Transverse section - U-shaped splayed vertebra, sac bulging through defect. (E) 3D surface rendering - nerve roots protruding into sac. (F) Ventriculomegaly + lemon sign.
Direct spinal signs:
- "U-shaped" or open vertebral arch on transverse section (splayed laminae)
- Cystic posterior lumbosacral mass - protruding sac
- Nerve roots visible within the sac (3D US)
Indirect cranial signs (almost pathognomonic):
- Lemon sign - frontal bone scalloping (biparietal diameter view)
- Banana sign - obliterated cisterna magna, curved/effaced cerebellum (Chiari II)
- Ventriculomegaly - secondary hydrocephalus
- Small posterior fossa, beaked tectum
Chiari II malformation is present between 19-25 weeks gestation in almost all MMC cases and may appear even in the first trimester. - Creasy & Resnik's MFM
5. AETIOLOGY & RISK FACTORS
Genetic
- Strong familial risk: A mother with one affected child has 20-50x increased risk for a subsequent NTD
- For an individual with spina bifida, the personal recurrence risk is 40x greater than normal
- Polygenic/multifactorial inheritance pattern
- Associations with mutations in folate metabolism pathways (MTHFR gene)
Environmental / Maternal Risk Factors
| Risk Factor | Evidence |
|---|---|
| Folate deficiency | Strongest modifiable risk; folic acid before day 28 is protective |
| Valproic acid / Carbamazepine | Folic acid antagonists; major teratogenic risk |
| Maternal diabetes (pre-gestational) | Well documented |
| Maternal obesity | Associated |
| Maternal fever/flu in early pregnancy | Associated |
| Young or advanced maternal age | Associated |
| Low socioeconomic status / low education | Associated |
| Maternal passive smoking | Associated |
| Maternal caffeine consumption | Some evidence |
| Previous miscarriage or birth defects | Associated |
| Higher birth order | Associated |
Only one-third of women take folic acid supplements as recommended. Government-mandated grain fortification with folic acid has resulted in a 20-50% decrease in NTD prevalence. - Campbell-Walsh-Wein Urology
6. CLINICAL PRESENTATION AT BIRTH
Visual Appearance
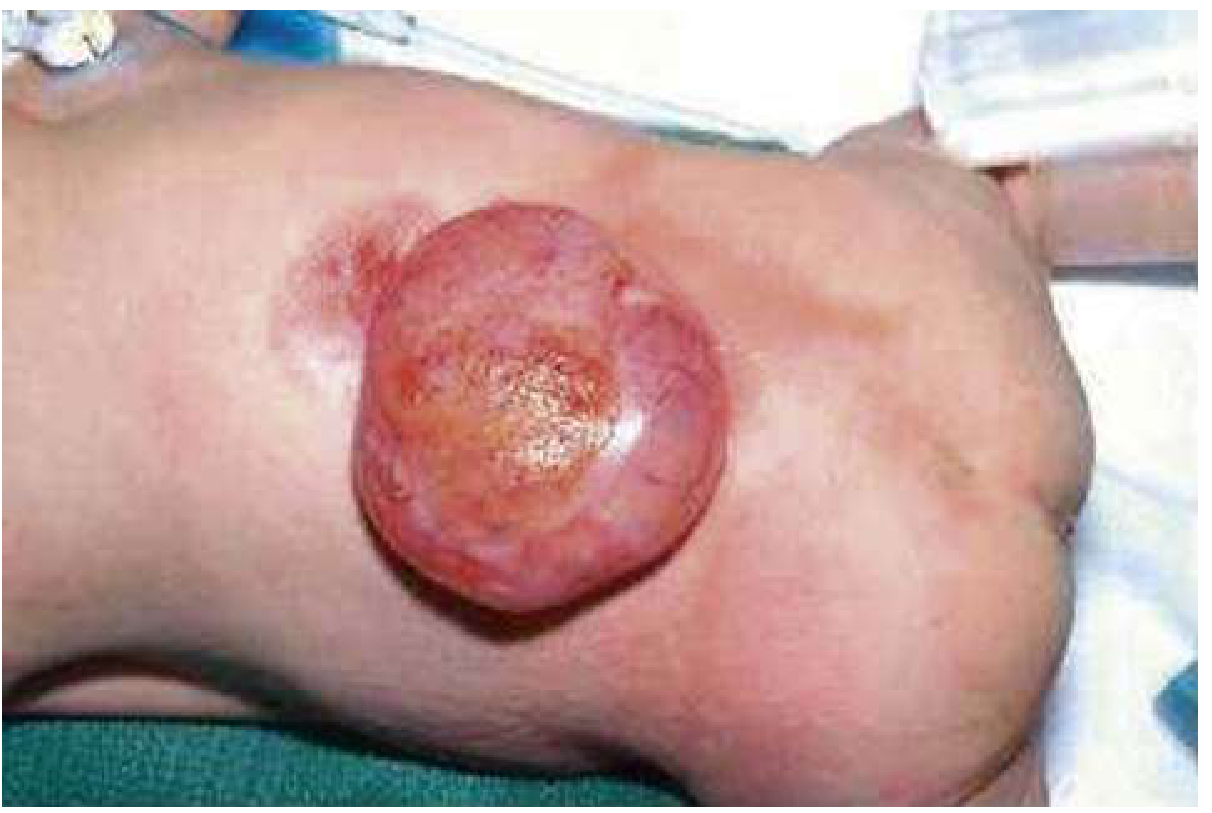
Rosen's Emergency Medicine - Fig. 159.3: Preoperative myelomeningocele, highlighting the obvious anatomic challenges and sensitivity of the exposed neural placode.
The newborn presents with a midline posterior lumbosacral sac covered by:
- Delicate, thin, often weeping/translucent membrane (NOT normal skin)
- May be covered by a thin membrane of arachnoid (partially skin-covered)
- Or completely open/ruptured - neural placode directly exposed (highest infection risk)
The defect may have ruptured in utero or during birth, but more often the covering is intact at delivery.
Level-Based Neurological Deficits - Critical Table
The neurological level is the highest functioning spinal segment. The bony vertebral level may differ from the neurological level by 1-3 vertebrae in either direction:
| Lesion Level | Motor Loss | Functional Ambulation |
|---|---|---|
| Thoracic | Total paralysis below thorax; flaccid lower limbs | Wheelchair dependent |
| High lumbar (L1-L2) | No knee extension, no hip extension; hip flexors may work | Ambulate with extensive bracing/crutches |
| Mid lumbar (L3-L4) | Quadriceps active; no ankle/foot movement; hip abductors weak | Community ambulation with AFOs possible |
| Low lumbar (L4-L5) | Ankle dorsiflexion preserved; plantar flexion weak; hip extension weak | Near-normal walking; AFOs may be needed |
| Sacral (S1-S2) | Plantar flexion and intrinsic foot muscles affected; perianal | Ambulate normally; bladder/bowel issues |
| Pure sacral (S3-S5) | Only bladder/bowel sphincters affected; legs escape | Normal walking; incontinence only |
The bony vertebral level often provides little or no clue to the exact neurological level or lesion produced. - Campbell-Walsh-Wein Urology
Neurological Examination at Birth
- Lower limb tone: Flaccid (LMN lesion)
- Lower limb movements: Absent (flaccid paralysis) or present only as reflex spinal arcs (NOT voluntary)
- Stroking the sac may elicit involuntary leg movements - these are spinal reflexes, NOT volitional
- Pinprick response: Absent over lumbosacral dermatomes
- Tendon reflexes: Absent
- Bladder: Continuous urinary dribbling (paralysed sphincter)
- Anus: Patulous anus; no anal wink
Head Examination - ALWAYS Examine
- Head circumference: Measure at birth - baseline for hydrocephalus monitoring
- Fontanelle: Bulging = elevated ICP from hydrocephalus
- Cranial sutures: May be split if hydrocephalus already present
- Sunset sign eyes: Downward gaze deviation in hydrocephalus
7. ASSOCIATED CONDITIONS - THE FULL PICTURE
A. Arnold-Chiari II Malformation (Universal)
Present in virtually ALL MMC patients. Components include:
- Downward herniation of cerebellar vermis + caudal brainstem through foramen magnum
- Small posterior fossa
- Kink/cervicomedullary kink in the medulla
- Hypoplastic pons
- Low-lying venous sinuses (low torcula)
- Abnormal corpus callosum (may be absent or thinned)
- Polymicrogyria (cortical malformation)
- Syringomyelia (syrinx in spinal cord)
Clinical consequences of Chiari II:
- Apnoea (central ± obstructive) - life-threatening in neonates
- Stridor - vocal cord dysfunction (cord paresis from vagal nerve stretching)
- Swallowing difficulties / aspiration
- Cranial nerve palsies (VI, VII, IX, X)
- Spasticity in upper limbs (if cervical cord compressed)
- Mortality: 35% in those with brainstem dysfunction at 5 years
B. Hydrocephalus
- >80% of MMC children require lifelong CSF diversion (Miller's Anesthesia)
- 81% have hydrocephalus requiring treatment (MFM data)
- 64-85% of lumbosacral MMC specifically require VP shunting
- 50% of children have shunt complications in the first year alone
- Mean IQ with VP shunting: 80 (low normal); lower than those not requiring shunts
- Chiari II is the mechanism: hindbrain herniation obstructs CSF through 4th ventricle
Hydrocephalus Management Options:
- Ventriculoperitoneal (VP) Shunt - traditional standard; risk of infection and repeated revision
- Endoscopic Third Ventriculostomy (ETV) + Choroid Plexus Cauterization (CPC):
- Manages hydrocephalus in >70% of MMC patients
- Avoids VP shunt
- Similar neurocognitive outcomes
- Preferred in some centres, especially resource-limited settings
- Campbell-Walsh-Wein Urology (Warf & Campbell, 2008)
C. Neurogenic Bladder (Universal)
- ALL MMC children have some degree of lower urinary tract dysfunction
- Bladder pattern depends on level - cannot be predicted by bony level alone
- Risk: silent hydronephrosis → renal scarring → renal failure
- Commence clean intermittent catheterisation (CIC) early (often from birth)
- Monitor with renal ultrasound and urodynamics
- Detrusor leak point pressure >40 cmH₂O = upper tract at risk
D. Neurogenic Bowel
- Anal sphincter paralysis → faecal incontinence and constipation
- Bowel programme: diet, laxatives, suppositories, Malone antegrade continence enema (MACE) procedure in older children
E. Orthopaedic Deformities
- Clubfoot (talipes equinovarus) - most common
- Hip dislocation (muscle imbalance)
- Progressive scoliosis - can be severe; eventually requires surgical fusion
- Kyphosis (gibbus deformity at the defect site)
- Knee contractures (flexion or extension)
- Calcaneus deformity
F. Skin / Pressure Ulcers
- Insensate skin below the neurological level
- Pressure sores over ischium, sacrum, heels - major source of morbidity
G. Latex Allergy
- MMC is associated with an elevated risk for latex allergy
- Mechanism: repeated mucosal exposure to latex (catheterisations, surgeries)
- Latex-free protocols must be established from birth for all MMC patients
- Rosen's Emergency Medicine
H. Cognitive & Neurodevelopmental
- Abnormal corpus callosum, polymicrogyria, small posterior fossa all contribute
- Lower cognitive function correlates with: higher lesion level, hydrocephalus requiring shunting
- Mean IQ ~80 (shunted patients)
- Non-verbal learning disabilities common
- Attention deficit, memory issues, maths difficulties despite relatively normal verbal IQ
I. Sexual Dysfunction
- Absent genital sensation and erectile dysfunction in males
- Female sexuality less affected but present
- Fertility may be preserved in females with lower-level lesions
8. NEONATAL MANAGEMENT - THE FIRST HOURS
Delivery Planning
- Caesarean section before onset of labour is recommended for fetuses with planned postnatal repair
- Improved motor function at 2 years observed in children born by elective C-section before labour vs. vaginal/emergency C-section
- Reason: minimising additional trauma to open neural elements during passage through birth canal
- Miller's Anesthesia 10e
Delivery Room - Immediate Actions
1. Position
Never place the MMC infant supine. Place prone or lateral to avoid any pressure on the defect.
- Rosen's Emergency Medicine
2. Protect the Placode
- Cover with sterile warm saline-soaked gauze (non-adherent)
- Enclose with plastic wrap (reduces heat and water loss from exposed neural tissue)
- Do NOT apply dry gauze directly to the placode
- Do NOT attempt to reduce or push the sac back
3. Temperature Management
- Neonates with MMC lose heat rapidly from the exposed sac surface
- Warm environment, plastic wrap, radiant warmer
4. Airway Assessment
- Assess for stridor, apnoea, swallowing difficulty - signs of Chiari II brainstem compression
- If present → urgent PICU admission, airway protection
- Central apnoea may require mechanical ventilation immediately
5. Intravenous Access & Antibiotics
- IV access (avoid lower limb veins if sensory-motor loss present - risk of undetected injury)
- IV antibiotics immediately - ampicillin + gentamicin (or local protocol)
- Prevents meningitis from bacterial colonisation of exposed neural tissue
Investigations - First 6 Hours
| Investigation | Purpose |
|---|---|
| Cranial ultrasound | Baseline ventricular size; identify hydrocephalus |
| MRI spine (urgent) | Level, extent, cord anatomy; pre-op planning |
| MRI brain | Chiari II characterisation, hydrocephalus, corpus callosum |
| Renal ultrasound | Baseline upper tract; identify hydronephrosis |
| Echo | Screen for CHD (~5%) |
| FBC, CRP, blood culture | Infection surveillance |
| U&E, creatinine | Baseline renal function |
| Urine output chart | Neurogenic bladder assessment |
| Karyotype | If dysmorphic / suspected chromosomal anomaly |
9. MULTIDISCIPLINARY TEAM - NON-NEGOTIABLE
Every MMC infant requires a named MDT from day 1:
| Specialist | Role |
|---|---|
| Neonatologist | Acute stabilisation, co-ordination |
| Paediatric neurosurgeon | Primary closure, VP shunt/ETV |
| Paediatric urologist | Neurogenic bladder, CIC, upper tract protection |
| Paediatric orthopaedic surgeon | Clubfoot, hip, spine |
| Paediatric physiotherapist | Mobility planning, splinting |
| Paediatric occupational therapist | Upper limb function, ADLs |
| Paediatric neurologist | Cognitive, epilepsy, Chiari management |
| Paediatric dietitian | Growth, nutrition (obesity common later) |
| Social work + family support | Parental counselling, community resources |
| Ophthalmology | Squint/VI nerve palsy from hydrocephalus |
10. SURGICAL MANAGEMENT
A. Postnatal Repair (Standard)
- Timing: within 24-72 hours of birth
- Goal: close the defect, prevent meningitis, preserve remaining neural function
- Procedure:
- Excise sac and devascularised tissue
- Identify and preserve neural placode
- Close neural tissue into a tube (neurorrhaphy)
- Watertight dural closure
- Fascial layer closure
- Skin closure (may require local flaps for large defects)
B. Fetal Surgery - The MOMS Trial (Landmark)
The Management of Myelomeningocele Study (MOMS) - NIH-funded multi-centre RCT:
Institutions: CHOP, UCSF, Vanderbilt, George Washington University
Population: 183 patients randomised (trial stopped early - prenatal surgery was so efficacious the safety monitoring board halted enrollment)
Inclusion criteria for prenatal surgery:
- MMC at L1-S1 level
- Evidence of hindbrain herniation
- Gestational age 19-26 weeks at randomisation
- Singleton pregnancy
- No major chromosomal anomaly
MOMS Trial Results:
| Outcome | Prenatal Repair | Postnatal Repair |
|---|---|---|
| Need for VP shunt at 12 months | 40% | 82-85% |
| Walking at 30 months | 42% | 21% |
| Chiari II reversal | Yes (improved) | No |
| Composite mental/motor score | Better | Worse |
| Maternal complications | Higher | Lower |
| Average gestational age at delivery | 34.1 weeks | 37.3 weeks |
| Delivery <30 weeks | 13% | 0% |
| Uterine scar dehiscence at delivery | ~33% | Rare |
Key takeaway: Prenatal repair delivers superior neurological outcomes but at significant maternal cost. 40% of prenatally repaired infants still needed VP shunting.
Maternal risks of open fetal surgery:
- Oligohydramnios
- Chorioamnionic separation
- Placental abruption
- Transfusion
- Spontaneous membrane rupture
- Preterm delivery (major risk)
Sabiston Textbook of Surgery; Mulholland & Greenfield's Surgery 7e; Campbell-Walsh-Wein Urology
C. Fetoscopic (Minimally Invasive) Fetal Repair
- Emerging technique at specialised centres
- Avoids maternal laparotomy and hysterotomy
- Less maternal morbidity
- Urinary tract functional outcomes similar to postnatal repair
- Higher fetal loss rates in early series; improving with experience
- Current focus of the post-MOMS systematic review (PMID 41342964, 2025)
11. NEUROGENIC BLADDER - DETAILED MANAGEMENT
This is a lifelong priority. The urological goal is to protect the upper urinary tracts (kidneys) while achieving social continence.
Urodynamic Patterns in MMC
The neurological level does NOT reliably predict bladder behaviour:
| Pattern | Features | Risk |
|---|---|---|
| Detrusor overactivity + high outlet resistance | High detrusor leak point pressure; VUR | High - upper tract damage |
| Detrusor underactivity + high outlet resistance | Retention; overflow incontinence | Moderate |
| Detrusor overactivity + low outlet resistance | Stress incontinence | Lower tract |
| Detrusor underactivity + low outlet resistance | Dribbling incontinence | Lower tract |
Management Ladder
- Clean Intermittent Catheterisation (CIC) - started in neonatal period
- Anticholinergic medications (oxybutynin) - reduce detrusor overactivity, protect upper tracts
- Alpha-blockers - reduce outlet resistance
- Botulinum toxin injection into detrusor - for refractory overactivity
- Bladder augmentation (ileocystoplasty) - for small, high-pressure bladder
- Continent catheterisable channel (Mitrofanoff procedure) - for patients unable to catheterise urethrally
- Malone antegrade continence enema (MACE) - for bowel management
12. ORTHOPAEDIC CONSIDERATIONS
Clubfoot (Talipes Equinovarus)
- Most common orthopaedic deformity
- Ponseti casting is first line (even in MMC - works well for flexible deformity)
- Minor posterior release may be needed
- Goal: plantigrade foot for orthosis fitting
Hip
- Dislocation common (especially L3-L4 level due to unopposed hip flexor/adductor activity)
- Management controversial - reduce only if ambulatory potential exists
- Hip reduction does NOT improve ambulation in high thoracic/lumbar lesions
Spine
- Kyphosis at birth (from bone defect + muscle imbalance) - may need kyphectomy at repair
- Scoliosis - progressive; onset often ~5-6 years; surgical fusion when curve >40-50°
- Combined anterior + posterior fusion improves pulmonary function
Ambulation Prognosis (Rule of Thumb)
- Sacral level: Community ambulation without aids
- Low lumbar (L4-L5): Community ambulation with AFOs
- Mid lumbar (L3): Ambulation with KAFOs and crutches (household ambulation)
- High lumbar/thoracic: Wheelchair primary; bracing physiotherapy possible
13. TETHERED CORD SYNDROME - CRITICAL LATE COMPLICATION
After MMC repair, the cord may re-tether at the repair site.
Signs of tethered cord:
- Progressive scoliosis or kyphosis
- Worsening lower limb strength or spasticity
- Worsening bladder/bowel function
- New back or leg pain
- Rapid deterioration around growth spurts
Diagnosis: MRI spine (low-lying conus, absent cord movement)
Treatment: Surgical cord detethering
14. OUTCOMES & PROGNOSIS
Mortality
- 14% mortality by 5 years overall
- 35% mortality by 5 years in those with Chiari II brainstem dysfunction symptoms
- With modern care (VP shunting + early closure), survival to adulthood is achievable
Long-term Function
| Outcome | Data |
|---|---|
| IQ >80 | >70% of survivors |
| Independent living as adults | Only 37% |
| Ambulation at 30 months (prenatal repair) | 42% |
| Ambulation at 30 months (postnatal repair) | 21% |
| Require lifelong VP shunting | >80% |
| Shunt complications in year 1 | ~50% |
| Mean IQ with VP shunt | ~80 |
Creasy & Resnik's MFM; Miller's Anesthesia 10e
15. PREVENTION
Folic Acid - Most Important Intervention
- 0.4-0.8 mg/day periconceptionally for all women of reproductive age (from at least 1 month before conception through first trimester)
- 4-5 mg/day for high-risk women (prior NTD pregnancy, on valproate/carbamazepine, diabetic, obese)
- Must be taken before day 28 (neural tube closes by day 28)
- Food fortification programmes: 20-50% reduction in NTD prevalence
- Limitation: folic acid supplementation does NOT eliminate MMC - cases continue to occur
Antiepileptic Drug Counselling
- Switch from valproate/carbamazepine to safer alternatives in women planning pregnancy
- If unavoidable, maximise folate supplementation and counsel about risks
16. QUICK EXAM PEARLS
- MMC = spinal cord + meninges herniated through vertebral defect → always neurological deficits
- 10x more common than meningocele
- Two-hit hypothesis: 1st hit = failed neural tube closure; 2nd hit = amniotic fluid toxicity + mechanical trauma → rationale for fetal surgery
- Chiari II present in virtually ALL MMC patients
- Never place MMC neonate supine - always prone/lateral
- Latex allergy risk - latex-free protocols from birth
- Folic acid prevents NTDs only if taken before 28 days post-conception
- MOMS trial: Prenatal repair halved VP shunt need (40% vs. 82%), doubled walking rate at 30 months (42% vs. 21%) - but major maternal risks
- Bony vertebral level ≠ neurological level - may differ by 1-3 vertebrae
- ETV + CPC manages hydrocephalus without VP shunt in >70% of cases
- VP shunt complications in year 1: ~50%
- Tethered cord must be excluded in any child with progressive decline after MMC repair
- Only 37% of MMC survivors live independently as adults
Sources:
- Campbell-Walsh-Wein Urology - Neurogenic Bladder in NTDs
- Creasy & Resnik's Maternal-Fetal Medicine - MMC Repair
- Mulholland & Greenfield's Surgery 7e - Myelomeningocele
- Sabiston Textbook of Surgery - MMC & Fetal Surgery
- Miller's Anesthesia 10e - Myelomeningocele
- Bradley & Daroff's Neurology in Clinical Practice
- Boron & Boulpaep Medical Physiology - Table 10-3
- Rosen's Emergency Medicine - Neonatal Emergencies
- Recent evidence: MOMS trial post-analysis systematic review (PMID 41342964, 2025) | Long-term neurosurgical management review (PMID 38700682, 2024)
Teache me as neonatatologist, paeditrcian in depth topic- Pyloric stenosis
Reading File
Finding Sources
Finding Sources
Searching PubMed
"pyloric stenosis" AND infant
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
I now have everything needed - multiple authoritative textbook sources, images of US donut sign, barium "string sign/double channel," and laparoscopic pyloromyotomy. Here is the comprehensive teaching:
Infantile Hypertrophic Pyloric Stenosis (IHPS) - In-Depth Teaching for Neonatologists & Paediatricians
1. DEFINITION
Infantile Hypertrophic Pyloric Stenosis (IHPS) is an acquired progressive hypertrophy and hyperplasia of the circular (and to a lesser degree longitudinal) muscle of the pylorus, resulting in narrowing and obstruction of the gastric outlet. It is not a true congenital defect - the pylorus is normal at birth and thickens postnatally.
"Pyloric stenosis is not a congenital disease." - Current Surgical Therapy 14e
2. EPIDEMIOLOGY
- Incidence: 1 in 300-900 live births (most quoted: ~1 in 300-400)
- Sex ratio: Males : Females = 4-5:1 (most important demographic fact)
- Males: 1/150 births
- Females: 1/750 births
- First-born male infants are at highest risk
- More common in Caucasians
- Premature infants - higher risk
- Young maternal age - associated risk factor
Robbins Pathology; Yamada's Textbook of Gastroenterology; Sabiston Textbook of Surgery
3. AETIOLOGY & RISK FACTORS
Genetic Factors
- Monozygotic twins: High concordance rate - strongest evidence for genetic basis
- Dizygotic twins and siblings: Increased risk, though less than monozygotic
- Family history: Incidence in general population ~0.2%; rises to ~6% in siblings of affected patients
- Maternal history: Children of affected mothers have higher risk than children of affected fathers
- Genome-wide association studies have linked several genes related to GI development - precise pathogenesis not yet understood
- Associated chromosomal conditions: Turner syndrome, Trisomy 18
Molecular / Pathological Mechanism
- Lack of nitric oxide synthase (NOS) in pyloric tissue - most plausible mechanism
- Nitric oxide is the inhibitory neurotransmitter responsible for pyloric relaxation
- Without NOS → failure of pyloric relaxation → progressive muscular hypertrophy from repeated forceful contractions
- Sabiston Textbook of Surgery; Yamada's GI Textbook
Environmental / Drug Associations
- Erythromycin exposure (oral or via breast milk) in the first 2 weeks of life - well-established risk
- Azithromycin - also associated
- Mechanism: macrolide antibiotics are motilin receptor agonists → stimulate strong gastric contractions → trigger pyloric hypertrophy
- Associated conditions: maternal myasthenia gravis, fetal rubella, phenylketonuria, Hirschsprung disease, oesophageal atresia, Smith-Lemli-Opitz syndrome, Cornelia de Lange syndrome
4. PATHOLOGY & PATHOPHYSIOLOGY
Gross Pathology
- Circular muscle layer hypertrophy (primary) + longitudinal muscle hypertrophy (secondary)
- Pyloric canal becomes markedly elongated and narrowed
- Pylorus appears as a firm, oval, pale "tumour" - the palpable "olive"
- Hypertrophic muscle bulges into the gastric antrum
Histology
- Hyperplasia of the muscularis propria (circular layer)
- Mucosal and submucosal oedema and inflammation exacerbate obstruction
- No primary neuronal abnormality on histology (unlike Hirschsprung disease)
Consequence of Obstruction
- Gastric outlet obstruction → forceful non-bilious vomiting (stomach content only - above the ampulla of Vater)
- Stomach becomes progressively distended and hypertrophied
- Repeated vomiting → loss of H⁺, Cl⁻, K⁺ → hypochloraemic, hypokalaemic metabolic alkalosis
- Progressive dehydration → weight loss, failure to thrive
5. METABOLIC DERANGEMENT - CRITICAL UNDERSTANDING
This is a mandatory mastery topic for the paediatrician/neonatologist.
How the Alkalosis Develops - Step by Step
| Step | Event | Metabolic Consequence |
|---|---|---|
| 1 | Persistent vomiting of gastric juice | Loss of HCl → loss of H⁺ and Cl⁻ |
| 2 | Hypochloraemia | Cl⁻ ↓ → renal HCO₃⁻ reabsorption increases (Cl⁻-HCO₃⁻ exchange) |
| 3 | Metabolic alkalosis develops | pH ↑, HCO₃⁻ ↑, pCO₂ ↑ (respiratory compensation) |
| 4 | Dehydration → aldosterone release | Na⁺ retention; K⁺ and H⁺ excretion → worsens hypokalaemia AND alkalosis |
| 5 | Paradoxical aciduria | Kidney excretes H⁺ despite systemic alkalosis - to conserve Na⁺; this is "paradoxical aciduria" |
| 6 | Late stage | Hyponatraemia may also occur |
Classic Biochemical Picture
| Parameter | Change | Value (approx) |
|---|---|---|
| pH | ↑ | >7.45 |
| Serum HCO₃⁻ | ↑ ↑ | Often >30 mEq/L |
| Serum Cl⁻ | ↓ ↓ | <90 mEq/L |
| Serum K⁺ | ↓ | <3.5 mEq/L |
| Serum Na⁺ | ↓ (variable) | May be low |
| pCO₂ | ↑ (compensation) | Raised |
| Urine pH | ↓ paradoxically | Acidic despite alkalosis |
Critical anaesthesia/surgical point: If bicarbonate >30 mEq/L, the infant has diminished respiratory drive and is at risk for postoperative apnoea and respiratory arrest under anaesthesia. Surgery must NEVER be performed until the metabolic alkalosis is fully corrected. - Current Surgical Therapy 14e; Sabiston
6. CLINICAL PRESENTATION - TIMELINE & SYMPTOMS
Typical Timeline
- Birth to 2 weeks: Normal - no symptoms (pylorus is normal at birth)
- 2-6 weeks: Onset of symptoms (peak 3-5 weeks)
- Range: 1 week to 3 months; rare after 3 months (unless premature infant)
- 20% of infants may develop symptoms before 3 weeks
Symptom Progression
Stage 1 (Early):
- Mild spitting / regurgitation after feeds
- Often misdiagnosed as GORD (GOR/reflux) or formula intolerance
- Infant remains hungry after vomiting and demands re-feeding immediately ("hungry vomiter")
Stage 2 (Classic):
- Non-bilious projectile vomiting - cardinal symptom
- Vomiting is NEVER bilious (obstruction is proximal to the ampulla of Vater)
- Projectile: may exit through nostrils as well as the mouth
- Occurs at the end of or shortly after feeding
- Not bile-stained; occasionally contains "coffee ground" material or small amounts of blood (from gastritis/oesophagitis)
- Visible gastric peristalsis - left-to-right wave across upper abdomen during or after feeding
- Infant still hungry and eagerly re-feeds after vomiting
Stage 3 (Late - Severely Unwell):
- Dehydration: sunken fontanelle, dry mucous membranes, poor skin turgor
- Wasted/marasmic appearance - loss of subcutaneous fat
- Decreased urine output (oliguria/anuria)
- Decreased stool output (constipation - nothing getting through pylorus)
- Jaundice - indirect hyperbilirubinaemia (related to volume depletion, caloric restriction, and possibly decreased hepatic glucuronyl transferase activity)
- Interest in feeding wanes as malnutrition progresses
7. PHYSICAL EXAMINATION - CLASSIC SIGNS
The Pyloric "Olive"
- Firm, ovoid, 1-2 cm mass palpable in the epigastrium or right of midline, below the liver edge
- Pathognomonic when present - no imaging required if palpated by experienced examiner
- Palpable in 70-90% of cases in experienced hands (now <30% due to earlier hospital presentation)
- Best felt:
- Immediately after vomiting (stomach decompressed, abdominal wall relaxed)
- With infant prone
- After NG tube decompression of stomach
- With infant calm (pain medications may help)
- During/after feeding (use finger to palpate while infant sucks)
- Location varies from umbilicus to epigastrium
Visible Peristaltic Waves
- Waves visible crossing upper abdomen left to right (stomach trying to force contents through stenosed pylorus)
- Best seen during feeding with infant undressed in good light
- Cradled in mother's left arm while being fed
Signs of Dehydration
- Sunken anterior fontanelle
- Sunken eyes
- Dry mucous membranes
- Reduced skin turgor
- Prolonged capillary refill
8. DIAGNOSIS & IMAGING
Approach to Diagnosis
- Classic presentation + palpable olive → diagnosis made clinically - no imaging required
- Typical presentation, no olive palpable → Ultrasound first line
- Equivocal US or atypical → Upper GI contrast study
Ultrasound - Gold Standard
Sensitivity and specificity up to 98-100%
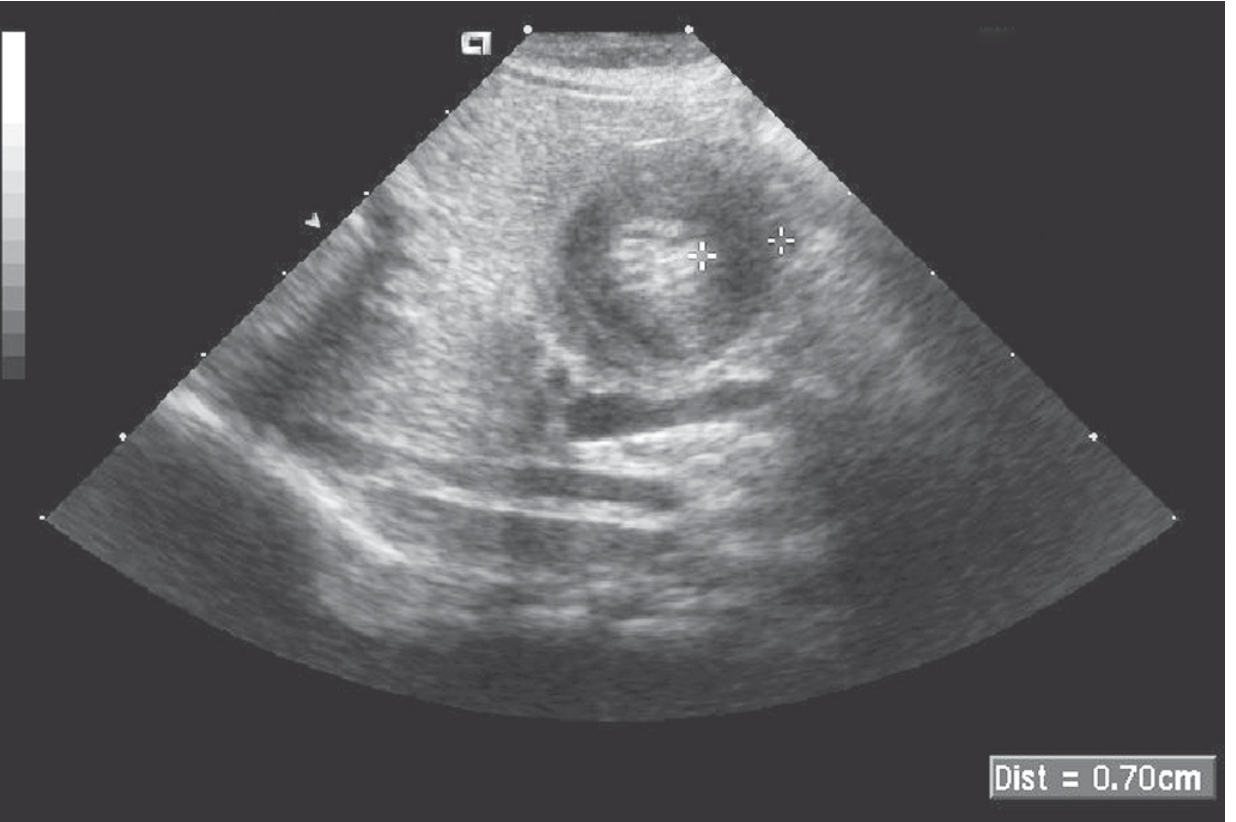
Sleisenger & Fordtran's GI and Liver Disease - Fig 49.9: The sonolucent "donut" of pyloric hypertrophy. Crossbars measure an abnormal 7mm muscle thickness.
US Diagnostic Criteria:
| Measurement | Diagnostic Threshold | Notes |
|---|---|---|
| Pyloric muscle thickness (PMT) | ≥3-4 mm | Most important; most sources use ≥3.5-4 mm |
| Pyloric channel length (PCL) | >15-17 mm | PCL >17mm only seen in HPS |
| Pyloric diameter | >13 mm | Supportive |
Additional US Findings (Grainger & Allison):
- "Shoulder sign": Hypertrophic muscle bulging into the gastric antrum
- "Nipple sign": Double-layered hypertrophic mucosa protruding into the stomach
- "Cervix sign": Overall appearance resembles the uterine cervix
- "Donut sign" (cross-section): Sonolucent ring of hypertrophied muscle around the echogenic mucosa
- Hypoechoic thickened pyloric muscle
- Hyperechoic elongated mucosa of the obstructed canal
- Exaggerated peristaltic waves visible in real-time
- Inability to see fluid passing through the pylorus during the entire examination
Volume depletion may affect US measurements - ensure adequate fluid resuscitation before the scan if possible. - Sleisenger & Fordtran
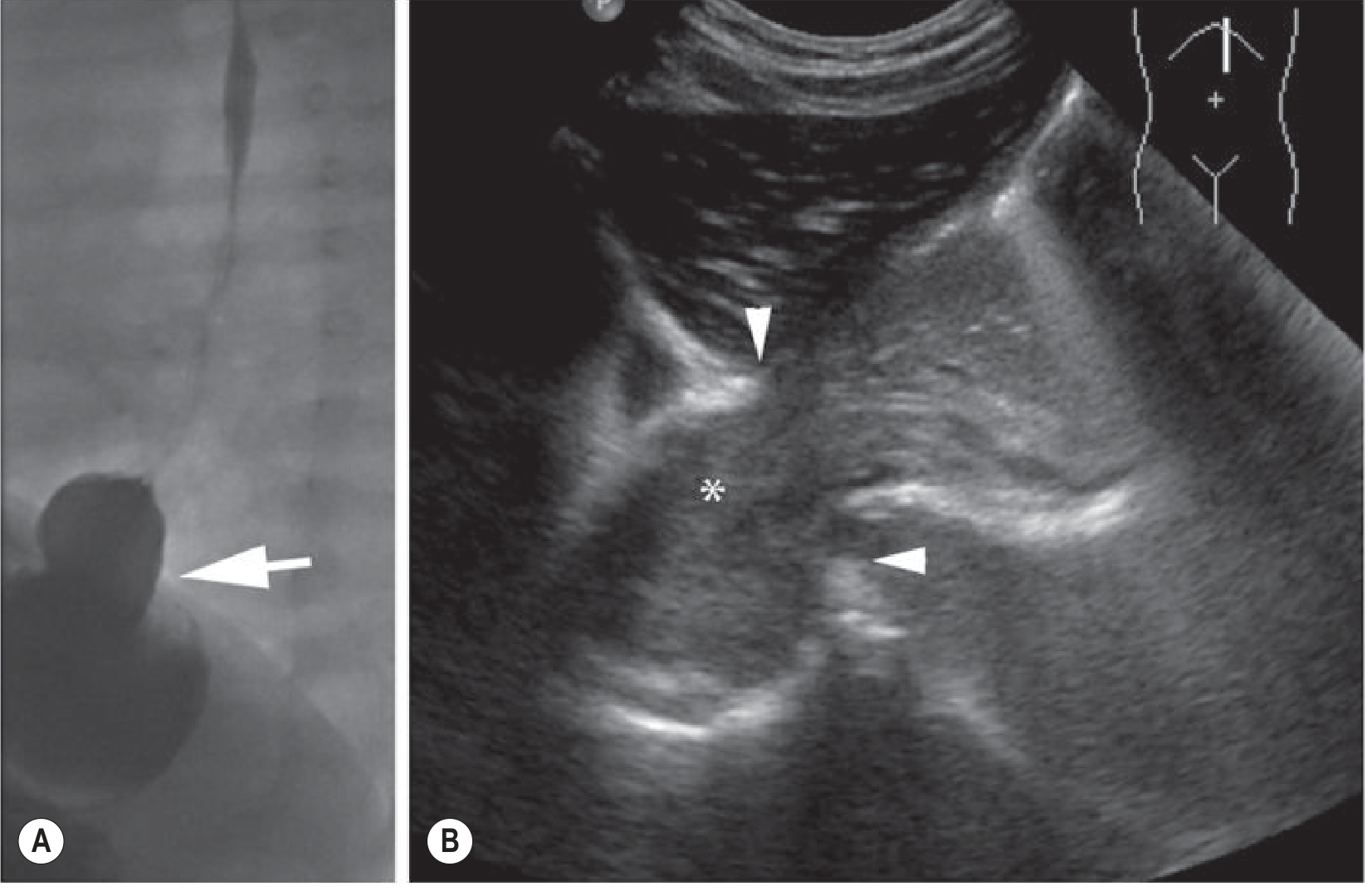
Grainger & Allison's Diagnostic Radiology - US of hypertrophic pyloric stenosis (A) and cross-section "donut" sign (B).
Upper GI Contrast Study (Barium)
Used when US is equivocal or for DDx including GORD/pylorospasm.
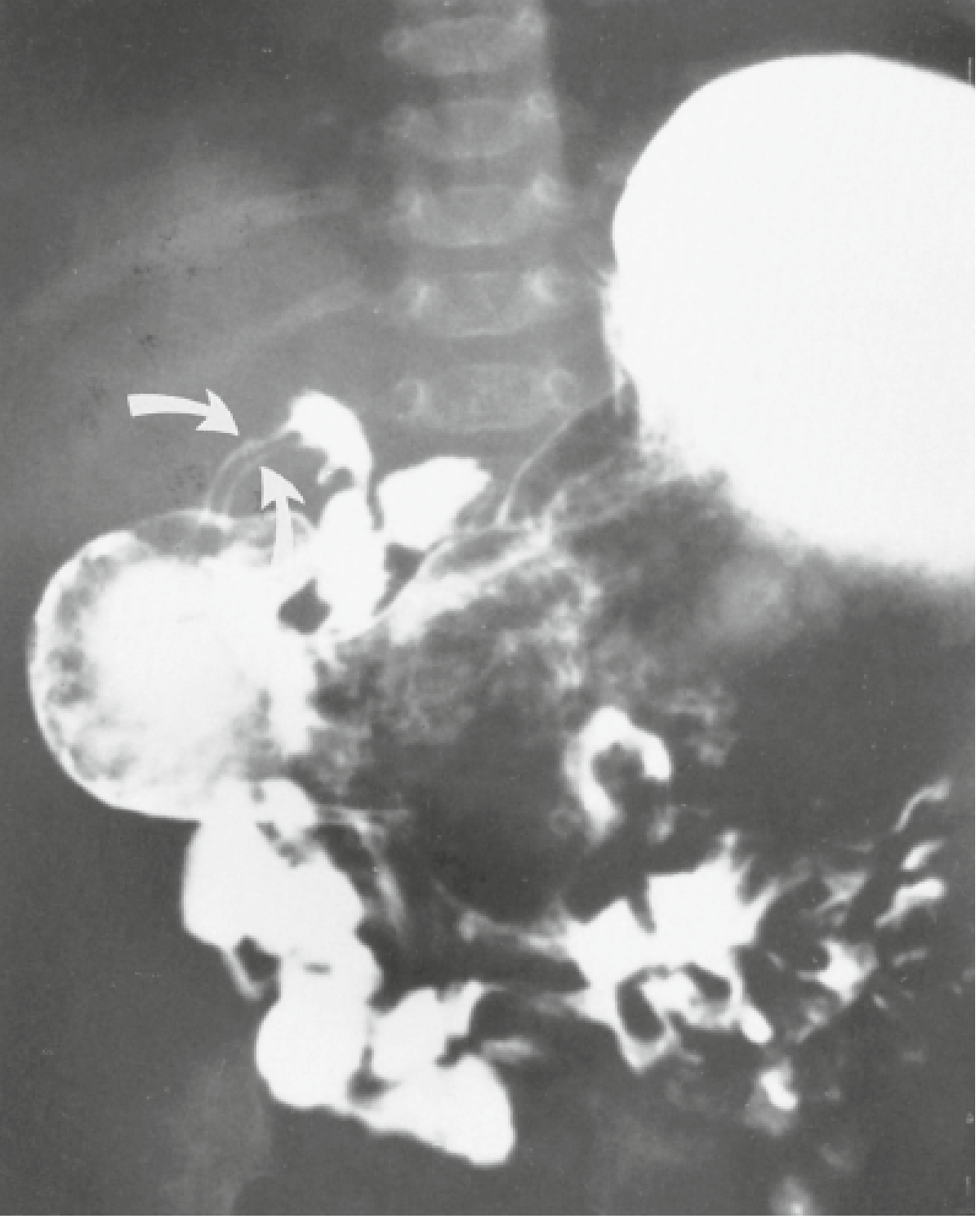
Yamada's Textbook of Gastroenterology - Fig 4.9: Barium contrast UGI series showing the long narrow "double channel" of the pylorus in HPS.
Barium Study Signs in HPS:
| Sign | Description |
|---|---|
| "String sign" | Thin line of barium forced through the narrow pyloric channel |
| "Double channel" / "tram-track" sign | Two parallel lines of barium in the elongated pyloric canal |
| "Mushroom/umbrella" sign | Compressed duodenal bulb with bulging pyloric mass |
| "Shoulder sign" | Pyloric mass indenting prepyloric antrum |
| "Caterpillar" sign | Visible gastric peristaltic waves on fluoroscopy |
| Markedly distended stomach | Gastric outlet obstruction |
| No passage of contrast | Absent or minimal transit through pylorus |
Barium study must be performed carefully with gastric contents aspirated first - infants are at highest risk for aspiration. - Sabiston Textbook of Surgery
Plain Abdominal X-Ray
- Enlarged gastric gas bubble - distended stomach
- Paucity of gas beyond the stomach (gas-less bowel distal to obstruction)
- Not diagnostic but supportive
Point-of-Care Ultrasound (POCUS)
- A 2023 meta-analysis (PMID 37722950) confirmed high diagnostic accuracy for POCUS in HPS when performed with vomiting + pyloric mass clinical features
9. DIFFERENTIAL DIAGNOSIS
| Condition | Key Distinguishing Features |
|---|---|
| GORD / GOR | Non-forceful, non-projectile; infant not wasted; US normal pylorus |
| Pylorospasm | Intermittent; US normal muscle thickness; resolves spontaneously |
| Overfeeding | Non-projectile; growth normal; no biochemical derangement |
| Duodenal atresia/stenosis | BILIOUS vomiting; "double bubble" on AXR; presents from day 1 |
| Malrotation/volvulus | BILIOUS vomiting; URGENT - mesenteric ischaemia |
| Antral web | Rare; endoscopy/UGI confirms |
| Adrenal insufficiency | Hyponatraemia, hyperkalaemia; pigmentation; female pseudohermaphroditism |
| Raised ICP | Neurological signs; projectile vomiting without hunger |
| Sepsis | Unwell; fever; other systemic signs |
| Intussusception | Older infant (3-12 months); "redcurrant jelly" stool; colicky pain |
Non-bilious vomiting = think above the ampulla of Vater (pylorus, duodenum proximal to bile duct) Bilious vomiting = ALWAYS investigate urgently = obstruction below the ampulla
10. INVESTIGATIONS - COMPLETE WORKUP
| Investigation | Findings in IHPS |
|---|---|
| Serum electrolytes | ↓Cl⁻, ↓K⁺, ↓Na⁺ (variable) |
| Venous blood gas | Metabolic alkalosis (↑pH, ↑HCO₃⁻, ↑pCO₂) |
| Serum bicarbonate | Often >30 mEq/L in severe cases |
| Serum chloride | Often <90 mEq/L |
| FBC | Haemoconcentration (raised haematocrit from dehydration) |
| Urea/creatinine | Elevated (pre-renal from dehydration) |
| Glucose | May be low (hypoglycaemia from poor intake) |
| Bilirubin | Indirect hyperbilirubinaemia |
| Urine dipstick | Paradoxical aciduria; concentrated |
| Abdominal US | Diagnostic (see above) |
11. MANAGEMENT
STEP 1: Resuscitation & Electrolyte Correction (MANDATORY BEFORE SURGERY)
Pyloric stenosis is NEVER a surgical emergency. It is a medical emergency first. - Miller's Anesthesia; Current Surgical Therapy 14e
Surgery must be deferred until dehydration and metabolic alkalosis are fully corrected.
Fluid Resuscitation Protocol:
Phase 1 - Volume Replacement (Bolus):
- Normal saline (0.9% NaCl): 20 mL/kg IV bolus (repeat if needed)
- Goal: restore intravascular volume, establish urine output
- Check urine output ≥1-2 mL/kg/hr before starting potassium
Phase 2 - Ongoing Replacement + Maintenance:
- Once urine output established:
- D5 ½ NS (0.45% NaCl) + KCl 20 mEq/L at 1.5x maintenance rate
- Electrolytes checked every 6-12 hours until normalised
Surgical GO Criteria (Endpoints):
| Parameter | Target |
|---|---|
| Serum Cl⁻ | ≥90-100 mEq/L |
| Serum HCO₃⁻ | ≤30 mEq/L |
| Serum K⁺ | ≥3.5 mEq/L |
| Urine output | Wet nappies (≥1-2 mL/kg/hr) |
| Serum Na⁺ | Normalised |
Correction may take 24-72 hours depending on severity. Do not rush to theatre. - Current Surgical Therapy 14e
NGT (Nasogastric Tube):
- Decompress the stomach to reduce aspiration risk
- Note: NGT can worsen electrolyte losses if left on free drainage - balance carefully
- Miller's Anesthesia notes: "Nasogastric tubes are not always used preoperatively as they may worsen the electrolyte imbalance"
- Before induction, suction stomach in supine and both lateral positions - removes ~98% of gastric contents
Other:
- NPO (nil by mouth)
- IV glucose to prevent hypoglycaemia
- Monitor urine output closely
STEP 2: Surgery - Ramstedt Pyloromyotomy
The Ramstedt-Fredet pyloromyotomy is the definitive treatment and is virtually always curative.
Principle of the Operation
Longitudinal incision through the anterior wall of the hypertrophied pyloric musculature down to (but NOT through) the submucosa. The muscle is then spread apart, allowing the mucosa to bulge freely. This relieves the obstruction without entering the GI lumen.
Approaches
| Approach | Details | Evidence |
|---|---|---|
| Laparoscopic (preferred) | 3-port: umbilical port + two 3mm stab incisions; most centres now use this | Shorter hospital stay, lower SSI rate |
| Open - right upper quadrant incision | Traditional Ramstedt incision | Equally effective |
| Open - periumbilical/supraumbilical | Cosmetically superior scar | Similar outcomes to RUQ |
A 2024 meta-analysis (PMID 38935193) compared umbilical vs. right upper transverse incisions - comparable outcomes.
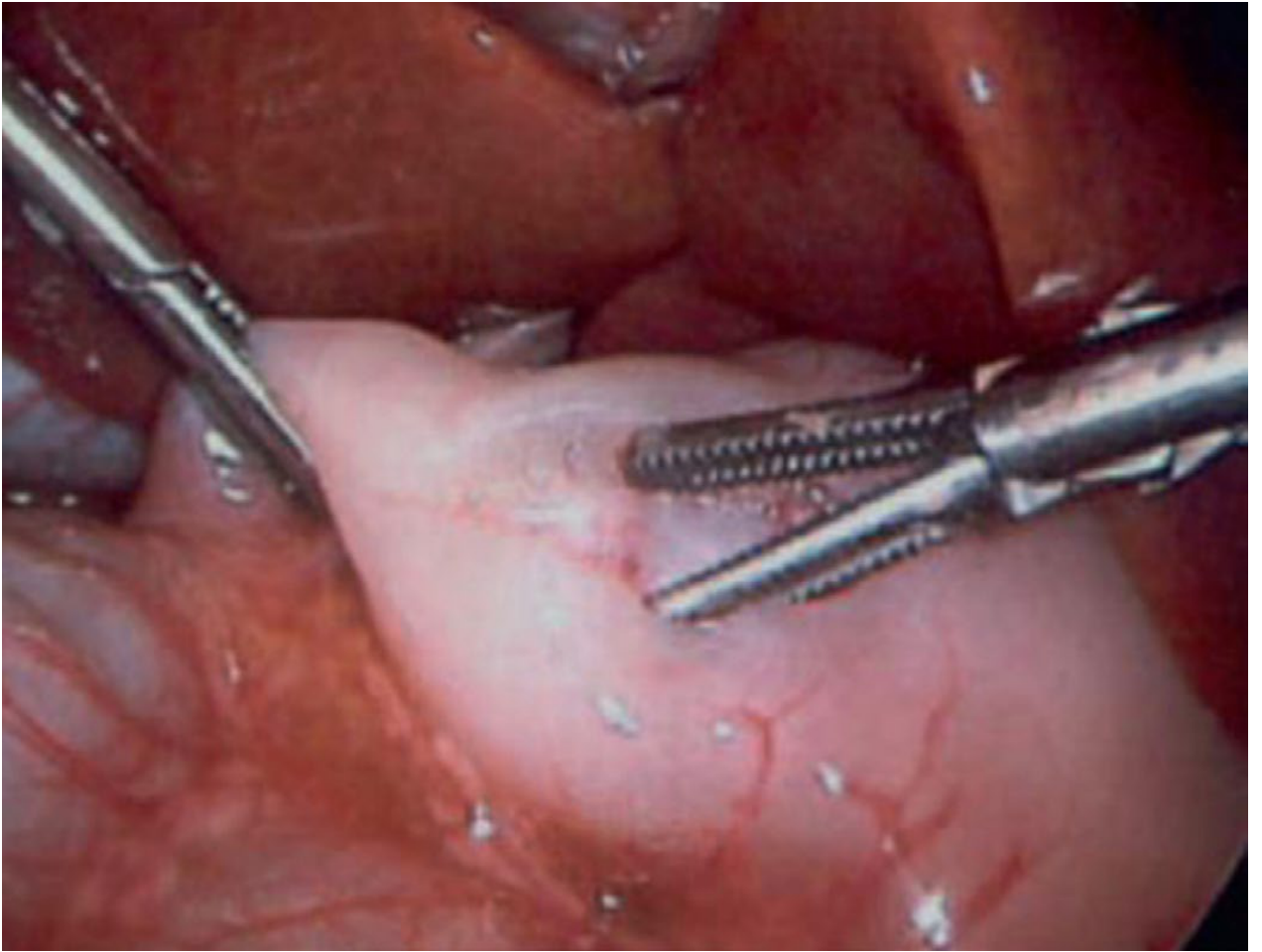
Sabiston Textbook of Surgery - Fig 117.6: Laparoscopic pyloromyotomy. A spreader is used to split the pyloric muscle. Intact mucosal bulging with independent muscle wall motion confirms complete myotomy.
Intraoperative Leak Test
- After myotomy: inject 30-60 mL of air through orogastric tube
- Confirm air passes through the pylorus into duodenum
- Confirm NO air leaks through mucosa (mucosal perforation excluded)
Anaesthetic Considerations (Critical for Neonatologist)
- Persistent metabolic alkalosis → depressed respiratory drive → postoperative apnoea risk
- Must correct CSF pH as well as plasma pH (CSF pH may lag several hours behind plasma normalisation)
- Rapid sequence intubation caution: cricoid pressure may distort anatomy; infants rapidly desaturate
- Pre-oxygenate; gentle mask ventilation before laryngoscopy is often necessary
- Atropine 0.02 mg/kg if succinylcholine 2 mg/kg used (prevents bradycardia)
- Post-op apnoea monitoring with pulse oximetry mandatory
- TAP block / local infiltration + paracetamol for analgesia
- Miller's Anesthesia 10e
STEP 3: Postoperative Feeding
- Mild vomiting in the first 24-48 hours postoperatively is normal and expected (gastric oedema resolves gradually)
- Ad lib feeding with initial volume limit (~60 mL) - current recommended approach
- Most infants tolerate full feeds within 24 hours
- Discharge: Most infants discharged within 36 hours after tolerating at least 2 consecutive feeds
- Current Surgical Therapy 14e
Prophylactic Antibiotics
A 2024 meta-analysis (PMID 39503899) examined preoperative prophylactic antibiotics for preventing SSI in IHPS - evidence still evolving; centre-dependent practice.
12. COMPLICATIONS
Intraoperative Complications
| Complication | Details | Management |
|---|---|---|
| Mucosal perforation | Most feared; occurs at duodenal end of myotomy | Repair perforation + close myotomy + rotate 180° + redo myotomy at new site; OR close + omentum buttress |
| Incomplete myotomy | Not extended far enough on stomach side | Return to OR for revision |
| Bleeding | Usually minor |
Postoperative Complications
| Complication | Details |
|---|---|
| Postoperative apnoea | Due to residual metabolic alkalosis - monitor with apnoea monitor + SpO₂ |
| Persistent vomiting >48h | Consider incomplete myotomy → return to OR |
| Wound infection | Laparoscopic approach has lower SSI rate |
| Aspiration | Prevent by gastric decompression before induction |
| Recurrent HPS | Rare; 2026 systematic review (PMID 41196118) - requires early re-operation |
13. PROGNOSIS
- Pyloromyotomy is virtually always curative
- After surgical correction, growth and development are completely normal
- Prognosis is excellent
- Mild vomiting may persist for days - expected; resolves as oedema settles
- The hypertrophied pylorus gradually involutes to normal size over weeks-months
- Recurrence after complete pyloromyotomy is extremely rare
14. SUMMARY TABLE - QUICK REFERENCE
| Feature | Details |
|---|---|
| Definition | Acquired hypertrophy of pyloric circular muscle → gastric outlet obstruction |
| Incidence | 1:300-900; M:F = 4-5:1 |
| Age at presentation | 2-6 weeks; range 1 week - 3 months |
| Vomiting character | Non-bilious, projectile, forceful; hungry after vomiting |
| Classic sign | Palpable "olive" in epigastrium; visible left-to-right peristalsis |
| Biochemistry | Hypochloraemic, hypokalaemic, metabolic alkalosis; paradoxical aciduria |
| Imaging | US: PMT ≥3-4 mm, canal length >15-17 mm; "donut sign" |
| Barium | "String sign," "double channel sign" |
| Surgery | Ramstedt pyloromyotomy - NEVER before electrolytes corrected |
| Pre-op target | HCO₃⁻ ≤30, Cl⁻ ≥90-100 mEq/L, wet nappies |
| Prognosis | Excellent - virtually always curative |
15. EXAM PEARLS
- HPS is NOT congenital - presents at 2-6 weeks, not at birth
- Vomiting is ALWAYS non-bilious (above ampulla of Vater)
- Infant is hungry after vomiting ("hungry vomiter") - classic distinguishing feature from other causes
- Paradoxical aciduria - kidney excretes acid despite alkalosis to conserve Na⁺ - pathognomonic for severe HPS
- Surgery is NEVER an emergency - always correct biochemistry first
- Bicarbonate >30 mEq/L = postoperative apnoea risk under anaesthesia - DO NOT operate
- If olive is palpated → no imaging needed - diagnosis made
- Bilious vomiting in a neonate = emergency (malrotation/volvulus until proven otherwise) - NOT HPS
- The macrolide connection: erythromycin in first 2 weeks of life increases HPS risk
- Laparoscopic pyloromyotomy is now preferred: shorter stay, lower infection rate
- Adequate myotomy = bulging mucosa + independent wall motion on both sides of the split
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease (Robbins Pathology)
- Sleisenger & Fordtran's Gastrointestinal and Liver Disease
- Yamada's Textbook of Gastroenterology 7e
- Sabiston Textbook of Surgery
- Current Surgical Therapy 14e
- Grainger & Allison's Diagnostic Radiology
- The Developing Human - Clinically Oriented Embryology (Moore & Persaud)
- Miller's Anesthesia 10e - Anaesthetic considerations in HPS
- Harriet Lane Handbook 23e (Johns Hopkins)
- Recent evidence: POCUS meta-analysis for HPS (PMID 37722950, 2023) | Umbilical vs RUQ incision meta-analysis (PMID 38935193, 2024) | Prophylactic antibiotics in HPS (PMID 39503899, 2024) | Recurrent HPS review (PMID 41196118, 2026)
Teache me as neonatatologist, paeditrcian in depth topic- Klinfetger syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Klinefelter syndrome" AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive multi-source content from Harrison's, Robbins, Campbell-Walsh, Harriet Lane, Langman's, Henry's Lab, and forensic medicine. Let me compile the full in-depth teaching response.
Klinefelter Syndrome (47,XXY) - In-Depth Teaching for Neonatologists & Paediatricians
1. DEFINITION & HISTORICAL NOTE
Klinefelter Syndrome (KS) is defined as male hypogonadism occurring when there are two or more X chromosomes and one or more Y chromosomes. The classic karyotype is 47,XXY.
First described in 1942 by Klinefelter, Reifenstein, and Albright as a syndrome of eunuchoidism, gynaecomastia, azoospermia, increased gonadotropin levels, and small firm testes. The 47,XXY karyotype was identified in 1959 by Jacobs and Strong.
- Campbell-Walsh-Wein Urology
It is the most common major abnormality of sexual development and the most common genetic cause of male infertility.
2. EPIDEMIOLOGY
| Parameter | Data |
|---|---|
| Incidence | ~1 in 500-660 live male births |
| % undiagnosed | ~75% of all cases never diagnosed |
| Diagnosis prepubertally (historical) | Only ~10% |
| Current trend | Noninvasive prenatal testing (NIPT) increasing early detection |
| Underdiagnosis rate | 25% diagnostic rate noted in Danish study |
Klinefelter syndrome is believed to be significantly underdiagnosed due to the wide range of phenotypic manifestations. Those with mild features are never seen by healthcare providers. - Robbins Pathology
3. GENETICS & KARYOTYPES
Classic Form: 47,XXY (90% of cases)
Robbins & Kumar Basic Pathology - Fig. 5.25: Clinical features and karyotype of Klinefelter syndrome.
Variants and Their Features
| Karyotype | Frequency | Notes |
|---|---|---|
| 47,XXY | ~90% | Classic Klinefelter |
| 46,XY / 47,XXY mosaic | ~10% | Milder phenotype; some fertility possible |
| 48,XXYY | Rare | More severe features; behavioural problems |
| 48,XXXY | Very rare | Intellectual disability more common |
| 49,XXXXY | Very rare | Most severe; intellectual disability, severe dysmorphism, joint hypermobility |
The physical attributes described for KS are quite variable - the only consistent finding is hypogonadism. - Robbins Pathology
4. MECHANISM OF NONDISJUNCTION - EMBRYOLOGY
What Goes Wrong
- Nondisjunction of sex chromosomes during meiosis in one of the parents
- An extra X chromosome is inherited
Who's to Blame?
- Maternal meiotic error (60%): Most common; extra X from mother
- Paternal meiotic error (40%): Extra X from father
Why Does the Extra X Cause Disease?
Two major pathological mechanisms:
- Aneuploidy effect: Increased gene dosage from supernumerary X chromosome (even with X-inactivation, ~15% of X-linked genes escape inactivation - these extra gene products are expressed)
- Hypogonadism: Seminiferous tubule failure → testosterone deficiency → loss of masculinisation
The Barr Body
- In 47,XXY males: one Barr body (sex chromatin body) is found in ~80% of nuclei
- Rule: number of Barr bodies = number of X chromosomes minus 1
- 47,XXY → 1 Barr body (same as normal female)
- 48,XXXY → 2 Barr bodies
- 49,XXXXY → 3 Barr bodies
- Langman's Medical Embryology
5. PATHOLOGY - TESTICULAR HISTOLOGY
Gross Findings
- Testes: firm, small - less than 3.5 cm in length
- Normal testes: 4-6 cm length; Klinefelter: significantly smaller
- Firmness = replacement of seminiferous tubules by hyaline material
Microscopic/Histological Findings
Three patterns described:
| Pattern | Features |
|---|---|
| Hyalinisation | Seminiferous tubules totally atrophied, replaced by pink, hyaline, collagenous "ghosts" - classic adult finding |
| Mixed | Apparently normal tubules interspersed with atrophic tubules |
| Primitive (embryonic) | All tubules are embryonic cords of cells - never developed a lumen or progressed to spermatogenesis |
Leydig Cell Appearances
- Leydig cells appear prominent and may resemble Leydig cell tumours (pseudo-hypertrophy)
- In reality: absolute Leydig cell volume is NOT increased - they appear prominent due to:
- Atrophy of adjacent germ cell compartment
- Crowding of tubules
- Elevated LH/gonadotropin concentrations
Robbins Pathology; Campbell-Walsh-Wein Urology
6. ENDOCRINOLOGY - HORMONAL PICTURE
| Hormone | Level | Explanation |
|---|---|---|
| FSH | ↑↑ Consistently elevated | Loss of inhibin from Sertoli cells → unopposed FSH rise |
| LH | ↑ Elevated | Loss of testosterone negative feedback |
| Testosterone | Low-normal to low | Leydig cell dysfunction |
| Oestradiol (Estradiol) | ↑ Elevated | Unknown exact mechanism; relative increase vs testosterone |
| SHBG | ↑ Elevated | Further reduces free testosterone |
| Inhibin B | ↓ Very low | Sertoli cell failure marker |
The ratio of oestrogens to testosterone determines the degree of feminisation (gynaecomastia) in individual patients. - Robbins Pathology
Hypergonadotropic hypogonadism is the endocrine pattern: primary testicular failure with elevated pituitary gonadotropins. - Forensic Medicine
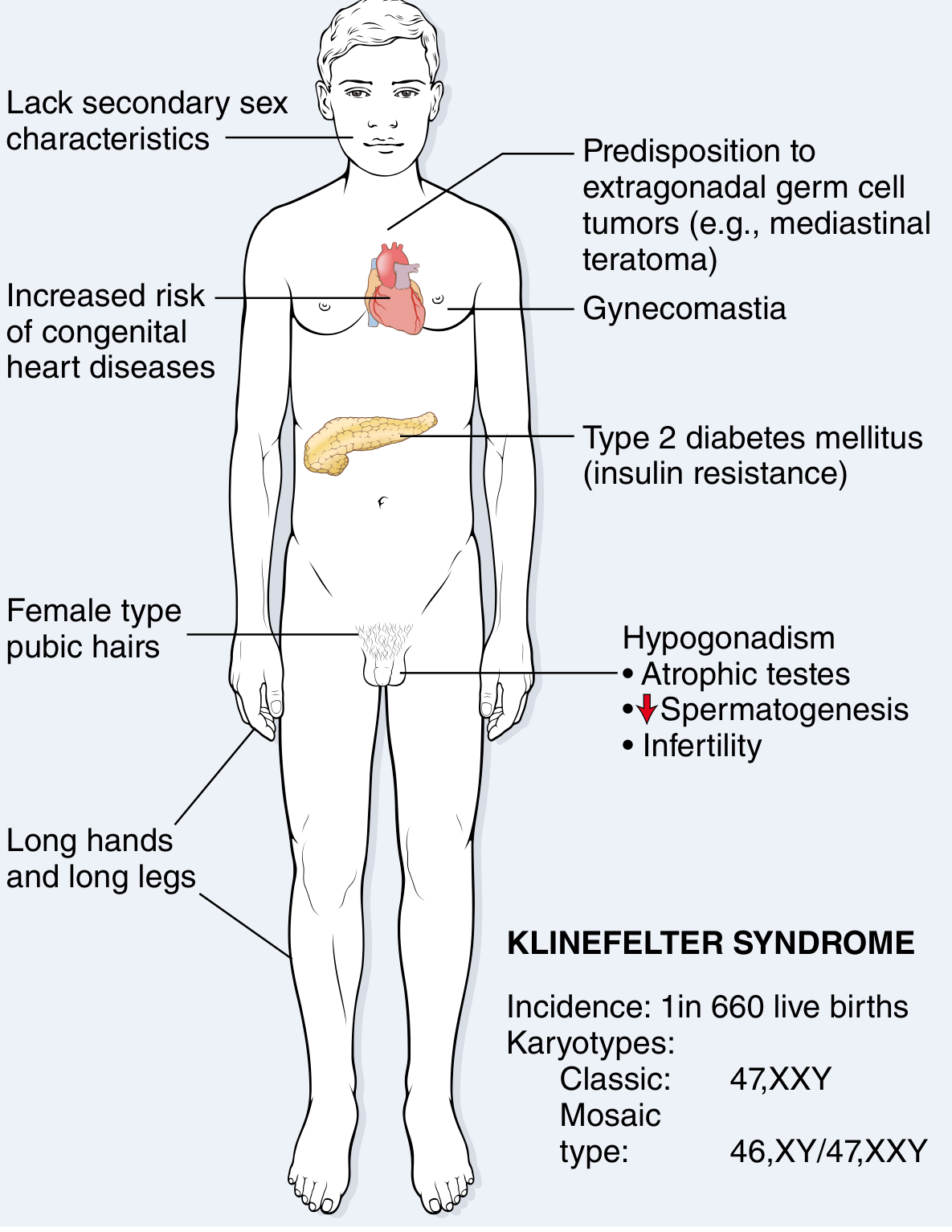
7. CLINICAL FEATURES - AGE BY AGE (PAEDIATRIC FOCUS)
A. Prenatal / Neonatal Detection
- NIPT (non-invasive prenatal testing): increasing rates of early identification
- Amniocentesis or chorionic villus sampling (CVS) - if suspected or incidental finding
- At birth: usually no obvious features distinguishing from normal males
- Occasionally: hypospadias or cryptorchidism - prompt karyotype
- Harriet Lane: "Primary hypogonadism which may present in infancy with hypospadias or cryptorchidism"
B. Infancy & Early Childhood
- Mostly NORMAL in appearance
- No obvious dysmorphic features
- Cryptorchidism (undescended testis) - important neonatal/paediatric finding
- May have slightly small penis
- Micropenis - associated with KS and other X polysomy syndromes
- Tall stature trend begins early (disproportionately long legs)
C. School Age (Pre-pubertal)
- Generally NOT identified by physical appearance
- Learning difficulties - most common reason for referral at this age
- Expressive language delay - cardinal finding in childhood
- Verbal IQ lower than performance IQ
- Reading and language comprehension difficulties
- Speech delay
- Behavioural problems: impulsivity, attention deficit, anxiety
- May have depressed verbal memory and processing speed deficits
- Taller than peers (long legs)
D. Puberty (Most Common Time of Diagnosis)
At puberty, the seminiferous tubules degenerate and hyalinise. This is when the syndrome becomes clinically obvious:
| Feature | Details |
|---|---|
| Gynaecomastia | Common pubertal development; can be marked; due to ↑ oestradiol : testosterone ratio |
| Small, firm testes | <3.5 cm length - pathognomonic |
| Failure of virilisation | Small penis, sparse/absent facial hair, absent/reduced body hair |
| Tall stature | Due to disproportionately long legs (present since childhood) |
| Eunuchoid body habitus | Increased lower segment (sole to pubis) > upper segment (pubis to crown) |
| Reduced muscle mass | Poor muscle development; fat distribution more female in pattern |
| Female-type pubic/axillary hair | Reduced terminal hair |
| Infertility (azoospermia) | Virtually universal in classic 47,XXY |
| Decreased libido | From androgen deficiency |
| Anxiety/depression | Neuropsychiatric component |
E. Adulthood (Classical Presentation / Delayed Diagnosis)
- Infertility - most common presentation in adults
- Postpubertal hypogonadism symptoms
- Metabolic complications (see section 8)
8. COMORBIDITIES & COMPLICATIONS - COMPLETE LIST
Metabolic / Endocrine
- Type 2 Diabetes Mellitus - increased incidence; due to insulin resistance / metabolic syndrome
- Metabolic syndrome - obesity, dyslipidaemia, insulin resistance
- Hypothyroidism - increased incidence
- Osteoporosis / Fractures - due to sex hormone imbalance (testosterone deficiency → reduced bone mineral density)
Cardiovascular
- Mitral valve prolapse - present in ~50% of adults with KS
- Atrial and ventricular septal defects - higher prevalence than general population
- Increased risk of thromboembolic disease
- Varicose veins
- Increased risk of leg ulcers
Oncological
- Extragonadal germ cell tumours (especially mediastinal teratoma): 20-30-fold increased risk
- Breast carcinoma: 8x the risk of normal males (due to gynaecomastia / elevated oestrogen)
- Leydig cell and Sertoli cell tumours - predisposition
- Routine testicular self-examination recommended postpubertally
Neuropsychiatric
- Verbal IQ deficit (specific - performance IQ relatively preserved)
- Reading and language comprehension difficulties
- Anxiety disorders
- Depression
- Attention difficulties (ADHD features)
- Reduced verbal memory
- Social communication difficulties (ASD-like features in some)
Autoimmune
- Systemic lupus erythematosus (SLE) - significantly increased incidence
- Autoimmune thyroid disease (Hashimoto's thyroiditis)
- Inflammatory bowel disease (some data)
Reproductive
- Azoospermia (virtually universal in classic 47,XXY)
- However: Testicular sperm extraction (TESE) + ICSI has achieved live births
- Meta-analysis: 43% live birth rate per ICSI cycle in KS patients
- But: lower proportion of normal embryos from KS patients (54%) vs controls (77%)
- Over 200 normal live births reported using ICSI without preimplantation genetic analysis
- Mosaic (46,XY/47,XXY): some natural fertility possible
Campbell-Walsh-Wein Urology; Robbins Pathology; Harrison's 22e
9. INTELLIGENCE & NEURODEVELOPMENT
- Intellectual disability is NOT a universal feature of classic 47,XXY
- Most patients have average to below-average cognitive abilities
- Specific deficit: Verbal skills, language, reading, comprehension
- Relatively preserved: Non-verbal/performance abilities
- The more X chromosomes → more likely to have intellectual disability:
- 47,XXY: usually average intelligence
- 48,XXXY: more likely cognitive impairment
- 49,XXXXY: most likely to have significant intellectual disability
- Langman's Medical Embryology
10. DIAGNOSIS
Who to Investigate?
- Infant with cryptorchidism + hypospadias
- Child with unexplained language/reading delay + tall stature
- Adolescent with gynaecomastia + small testes
- Adult male with azoospermia/infertility
- Adult male with hypogonadism
- Incidentally found on NIPT or amniocentesis
- Any male with learning difficulties + tall/eunuchoid habitus
Diagnostic Tests
| Test | Finding in KS |
|---|---|
| Karyotype (blood lymphocytes) | 47,XXY (gold standard) - 90% of cases |
| Buccal smear for Barr body | 1 Barr body (= sex chromatin positive) - rapid screening (now largely replaced by karyotype) |
| FISH (Fluorescence in situ hybridisation) | Rapid detection of X/Y copy number |
| Microarray / SNP array | Detects mosaicism; more sensitive than standard karyotype |
| NIPT (prenatal) | Can detect sex chromosome aneuploidy from maternal blood |
| Amniocentesis / CVS | Karyotype from fetal cells - definitive prenatal diagnosis |
Hormonal Investigations
| Test | Expected Result in KS (post-pubertal) |
|---|---|
| Serum FSH | ↑↑ (markedly elevated) |
| Serum LH | ↑ Elevated |
| Serum Testosterone | Low to low-normal |
| Serum Oestradiol | ↑ Elevated |
| Serum Inhibin B | ↓ Very low/undetectable |
| Semen analysis | Azoospermia |
| Bone mineral density (DEXA) | Reduced (osteopenia/osteoporosis) |
11. MANAGEMENT - MULTIDISCIPLINARY & AGE-SPECIFIC
A. Neonatal/Infant
- If detected on NIPT → genetic counselling for parents
- Confirm with karyotype at birth
- Examine genitalia: document testicular size, position, penile length
- If cryptorchidism → referral to paediatric urology (orchidopexy typically at 6-12 months)
- Screen for hypospadias
- No testosterone therapy needed at this stage
- Inform and support parents - prognosis is generally good
B. Early Childhood (2-8 years)
- Developmental surveillance - speech/language, reading, cognition
- Early referral to speech-language therapist if expressive language delay detected
- Educational psychology assessment
- Educational support - targeted literacy interventions
- Psychological support / counselling
C. Pre-puberty / Puberty (10-14 years)
- Monitor growth and pubertal development
- Testicular volume - document by Prader orchidometer at each visit
- If KS testes: small and firm; minimal growth at Tanner 2
- Testosterone therapy - initiate at expected time of puberty (11-12 years):
- Goal: induce/support masculinisation
- Route: IM testosterone esters (monthly), transdermal gel (daily), subcutaneous pellets
- Start low, titrate upward to mimic normal pubertal progression
- Harriet Lane Handbook: "Testosterone therapy is indicated at puberty for hypergonadotropic hypogonadism"
- Monitor LH, FSH, testosterone, bone age, DEXA
D. Testosterone Replacement Therapy - Details
| Aspect | Details |
|---|---|
| Indication | Hypergonadotropic hypogonadism confirmed |
| Goal | Normalise testosterone, promote virilisation, prevent osteoporosis, improve metabolic profile, improve mood/energy |
| Age to start | At onset of puberty (~11-12 years) |
| Route options | IM injections (every 2-4 weeks), transdermal gel, topical, subcutaneous implants |
| Monitoring | Testosterone levels, LH/FSH, haematocrit, DEXA, lipids, bone age |
| Does NOT | Restore fertility (testes remain dysfunctional; sperm production does not restart) |
| Gynaecomastia | May require surgical reduction (mastectomy) if persistent/severe |
E. Fertility - Advanced Options
- Testicular Sperm Extraction (TESE) + ICSI is the only path to biological parenthood
- Best undertaken before testosterone replacement (or after a prolonged washout) - exogenous testosterone suppresses residual spermatogenesis
- Window: adolescence/young adulthood - germ cell depletion worsens with age
- Outcome: 43% live birth rate per ICSI cycle
- Option: cryopreserve sperm extracted during adolescence for later use
- Preimplantation genetic testing (PGT): Optional; does not clearly improve outcomes
- Mosaic KS: higher chance of natural sperm production; fertility counselling earlier
F. Long-Term Monitoring & Comorbidity Surveillance
| System | Monitoring |
|---|---|
| Bone health | DEXA at diagnosis; repeat periodically; calcium + vitamin D supplementation |
| Cardiovascular | Echocardiogram (MVP, ASD/VSD); lipid profile; blood pressure; glucose |
| Metabolic | Fasting glucose/HbA1c annually; weight monitoring; lipids |
| Oncological | Testicular self-examination (monthly); breast self-examination; low threshold for imaging |
| Endocrine | Testosterone, LH, FSH, thyroid function, HbA1c |
| Neurodevelopmental | Ongoing educational support; psychological input |
| Mental health | Screen for anxiety, depression; psychosocial support |
G. Gynecomastia Management
- If mild: reassurance; may partially improve with testosterone therapy
- If persistent or cosmetically significant → surgical mastectomy / breast reduction
- Monitor for breast carcinoma (8x risk vs normal males)
12. VARIANTS - DETAILED (PAEDIATRIC RELEVANCE)
46,XY / 47,XXY Mosaicism
- Milder phenotype than classic KS
- Some natural fertility possible (depends on ratio of XY to XXY cells)
- Higher XY proportion → more phenotypically normal
- More likely to be identified during infertility workup
48,XXYY
- Taller stature
- More pronounced hypogonadism
- Behavioural problems more prominent
- Higher rates of ADHD, autism spectrum features
- Learning disabilities more severe
48,XXXY
- Cognitive impairment more common
- Severe hypogonadism
- Distinctive facies (epicanthal folds, hypertelorism)
- Joint hypermobility
49,XXXXY
- Most severe variant
- Significant intellectual disability
- Severe radioulnar synostosis
- Hypertelorism, epicanthal folds
- Cardiac defects
- Hypoplastic genitalia
- Clinodactyly
13. DIFFERENTIAL DIAGNOSIS
| Condition | Distinguishing Features |
|---|---|
| Kallmann syndrome | Hypogonadotropic hypogonadism (LH/FSH low, not elevated); anosmia; normal karyotype 46,XY |
| Constitutional delay of puberty | LH/FSH normal or low; bone age delayed; spontaneous progression; normal karyotype |
| Idiopathic hypogonadotropic hypogonadism | Low LH/FSH; normal karyotype |
| True hermaphroditism (ovotesticular DSD) | Karyotype variable; ovarian tissue present |
| 5α-reductase deficiency | 46,XY; normal testosterone; low DHT; ambiguous genitalia |
| Androgen insensitivity syndrome | 46,XY; female or ambiguous phenotype; testosterone normal/high |
Key distinguishing feature of KS: Hypergonadotropic (high LH, high FSH) hypogonadism + 47,XXY karyotype. - Forensic Medicine; Robbins
14. PAEDIATRIC PRESENTATION TIMELINE SUMMARY
| Age | Presentation | Action |
|---|---|---|
| Prenatal | NIPT/amniocentesis finding | Genetic counselling; confirm karyotype at birth |
| Neonatal | Cryptorchidism, hypospadias, micropenis | Karyotype; paediatric urology referral |
| Infant | Usually normal | If cryptorchid → orchidopexy 6-12 months |
| Toddler/preschool | Speech/language delay | SLT referral; developmental assessment |
| School age | Learning difficulties, reading delay, tall | Educational psychology; karyotype if not done |
| Puberty | Gynaecomastia, small firm testes, poor virilisation | Karyotype, hormones; start testosterone |
| Adolescent/adult | Azoospermia, infertility | TESE/ICSI; testosterone therapy |
15. QUICK EXAM PEARLS
- KS = 47,XXY - most common sex chromosome abnormality causing male infertility
- Most common genetic cause of male infertility (even ahead of Y chromosome microdeletions)
- ~75% of KS cases are never diagnosed in their lifetime
- Only consistent finding = hypogonadism
- Testes: small, firm (<3.5 cm) - key physical sign
- Histology: hyalinised seminiferous tubules + apparent Leydig cell prominence
- Hormones: FSH markedly elevated (most consistent) + LH ↑ + testosterone low + oestradiol ↑
- Barr body present (one, same as normal female) - nuclear sexing is "female" despite male anatomy
- Nondisjunction: 60% maternal, 40% paternal
- More X chromosomes → more intellectual disability: 47,XXY (mild/none) → 49,XXXXY (severe)
- Pubertal presentation: gynaecomastia + small firm testes + eunuchoid habitus
- Childhood presentation: expressive language delay + learning difficulties + tall stature
- Neonatal presentation: cryptorchidism ± hypospadias → always karyotype
- Testosterone therapy at puberty = standard of care; does NOT restore fertility
- TESE + ICSI: only fertility option; 43% live birth rate per cycle
- Breast cancer risk: 8x normal males
- Extragonadal germ cell tumours: 20-30x normal (especially mediastinal teratoma)
- Mitral valve prolapse in ~50% of adults
- KS is never a surgical emergency but has lifelong multisystem implications
Sources:
- Harrison's Principles of Internal Medicine 22e (2025)
- Robbins & Kumar Basic Pathology (Robbins Pathology)
- Robbins, Cotran & Kumar Pathologic Basis of Disease
- Campbell-Walsh-Wein Urology - Klinefelter Syndrome and Variants
- Harriet Lane Handbook 23e (Johns Hopkins Hospital)
- Langman's Medical Embryology
- Henry's Clinical Diagnosis and Management by Laboratory Methods
- Forensic Medicine & Toxicology 36th Ed (2026)
- Recent evidence: Blackburn J et al, Klinefelter Syndrome: A Review, Clin Endocrinol 2025 (PMID 39806878) | Mehmet B et al, Update on Physical, Psychological, and Quality of Life Management in KS, JCEM 2025 (PMID 40314150)
Teache me as neonatatologist, paeditrcian in depth topic- Turner syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Turner syndrome" AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive multi-source material with clinical photos and diagrams. Here is the full in-depth teaching:
Turner Syndrome - In-Depth Teaching for Neonatologists & Paediatricians
1. DEFINITION
Turner Syndrome (TS) is characterised by primary hypogonadism in phenotypic females resulting from complete or partial monosomy of the X chromosome. The classic karyotype is 45,X (monosomy X).
It is the most common sex chromosome abnormality in females and the only human viable monosomy.
2. EPIDEMIOLOGY
| Parameter | Data |
|---|---|
| Incidence in live female births | ~1 in 2,000-2,500 |
| 45,X karyotype | Most common cytogenetic abnormality in spontaneously aborted fetuses |
| Accounts for | ~18% of all chromosomally abnormal spontaneous abortions |
| Survival of 45,X embryos | Only ~1% of monosomy X embryos survive to birth |
| Diagnosed | ~50% are never formally diagnosed |
| 45,X → spontaneous abortion | ~98% are aborted spontaneously |
The 45,X karyotype is the single most frequent chromosomal disorder in humans, but most affected fetuses are aborted spontaneously early in pregnancy. Any short, slowly growing, sexually infantile girl has Turner syndrome until proved otherwise - the disorder is so prevalent (~1 in 2,500). - Berek & Novak's Gynecology
3. GENETICS & KARYOTYPES
Classic Form: 45,X (57% of cases)
Robbins & Kumar Basic Pathology - FIG. 4.20: Clinical features and karyotypes of Turner syndrome.
Full Karyotypic Classification
| Karyotype | Frequency | Clinical Notes |
|---|---|---|
| 45,X | ~57% | Classic full phenotype; most severe |
| 46,X,i(Xq10) - isochromosome of long arm | ~14% | Loss of short arm → full TS phenotype; highest hypothyroidism rate (50%) |
| 46,X,r(X) - ring chromosome | Rare | Variable phenotype |
| 46,X,del(Xp) - short arm deletion | Rare | Full phenotype (Xp deletion → TS stigmata) |
| 46,X,del(Xq) - long arm deletion | Rare | Short stature + ovarian failure; fewer somatic features if distal Xq |
| 45,X / 46,XX mosaicism | ~15-20% | Milder phenotype; may present only with primary amenorrhoea; rare spontaneous fertility |
| 45,X / 46,XY mosaicism | ~5-10% | Y-line → risk of gonadoblastoma; variable genitalia |
| 45,X / 47,XXX | Rare | Variable |
With more sensitive techniques, mosaicism prevalence rises to 75%; conventional cytogenetics detects ~30%. - Robbins Pathology (Cotran)
Which X is Missing?
- Paternal gamete (sperm) is missing in ~75% of cases (i.e., the father failed to contribute a sex chromosome)
- Error occurs predominantly in paternal gametogenesis
- Maternal meiotic error accounts for the remainder
- Langman's Medical Embryology
4. PATHOGENESIS - WHY DOES MISSING ONE X CAUSE ALL THIS?
X Inactivation and Gene Dosage
- Normally, one X chromosome is inactivated (Lyon hypothesis - Barr body)
- However, ~20% of X-linked genes escape X inactivation and need to be expressed from BOTH X chromosomes
- In 45,X: these genes are hemizygous → haploinsufficiency → TS features
SHOX Gene - Key for Short Stature
- SHOX (Short Stature Homeobox-containing gene) is located on the pseudoautosomal region of Xp22
- Escapes X inactivation → normally present in 2 copies
- In 45,X: only 1 copy → SHOX haploinsufficiency → short stature + skeletal defects
- Deletions distal to Xq21 do NOT affect stature (SHOX not involved)
- Loss of Xp → full TS phenotype (including SHOX)
- Loss of distal Xq → short stature + ovarian failure only
Berek & Novak's Gynecology
Ovarian Failure Mechanism
- Patients with Turner syndrome initially have normal ovarian development in utero
- After birth: accelerated follicular atresia → premature ovarian insufficiency
- Result: fibrotic, white streak ovaries ("streak gonads") devoid of follicles
- Histology: white fibrous stroma, no primordial follicles
- Berek & Novak's Gynecology
5. CLINICAL FEATURES - AGE-BY-AGE (PAEDIATRIC FOCUS)
Langman's Medical Embryology - Patient with Turner syndrome. A: At birth - loose skin at posterior neck (cystic hygroma remnant), short neck. B/C: Lymphoedema of hand and foot. D: At 6 years - webbed neck, widely spaced nipples.
A. Prenatal Detection
- NIPT (non-invasive prenatal testing): detects sex chromosome monosomy
- Ultrasound findings suggesting TS:
- Cystic hygroma (large fluid-filled posterior neck mass) - most characteristic
- Nuchal translucency ↑↑
- Fetal hydrops
- Cardiac defects (coarctation of aorta, bicuspid aortic valve)
- Horseshoe kidney
- Intrauterine growth restriction (IUGR)
- Brachycephaly
- Amniocentesis or CVS → karyotype confirmation
B. Neonatal Presentation
| Feature | Description |
|---|---|
| Lymphoedema of dorsum of hands and feet | Most characteristic neonatal finding; due to lymphatic obstruction |
| Loose skin at posterior neck | Remnant of in-utero cystic hygroma |
| Low birth weight / IUGR | Intrauterine growth restriction |
| Low posterior hairline | Visible from birth |
| Congenital heart disease | Detected by cardiac examination + echo |
| Short neck | Subtle at birth |
| Malformed ears | Low-set ears |
| Coarctation of aorta | Differential femoral vs brachial pulses; radio-femoral delay |
| Webbed neck | Pterygium colli - may not be obvious until older |
If a newborn girl has lymphoedema of hands/feet → Turner syndrome until proven otherwise - refer for karyotype.
C. Infancy & Early Childhood
- Lymphoedema subsides but leaves bilateral neck webbing and posterior loose skin
- Growth begins to slow from 2nd-3rd year of life
- Recurrent acute otitis media (AOM) and otitis media with effusion (OME) - very common
- Often the first presentation to a clinician before TS is diagnosed
- Eustachian tube dysfunction related to mid-facial anatomy
- Scott-Brown's Otorhinolaryngology: "The vast majority of girls with TS present to otolaryngologists in their early years with recurrent AOM and OME... for many this occurs long before the diagnosis of TS has been made"
- Feeding difficulties in infancy (poor suck, high-arched palate)
- Multiple pigmented naevi
- Short stature becomes increasingly apparent
D. School Age (Pre-pubertal)
| Feature | Details |
|---|---|
| Short stature | Below 3rd centile; typically >2 SD below mean; final untreated height rarely >150 cm |
| Webbed neck / low posterior hairline | Increasingly apparent |
| Cubitus valgus | Increased carrying angle of the elbows |
| Shield chest | Broad chest with widely spaced nipples |
| Short 4th metacarpal | Positive metacarpal sign (dimple when 4th knuckle extended) |
| High-arched palate | Present |
| Multiple pigmented naevi | Scattered |
| Otitis media | Recurrent; may develop chronic OME and conductive hearing loss |
| Space-form blindness | Difficulty appreciating shapes and spatial relationships |
| Learning difficulties | Visuospatial, maths, attention; normal verbal IQ |
| Alopecia areata | Increased prevalence |
E. Pubertal Failure (Most Common Time of Diagnosis in Previously Undiagnosed Girls)
| Feature | Details |
|---|---|
| No spontaneous breast development (failure of thelarche) | Most striking pubertal finding |
| Primary amenorrhoea | No menstruation |
| Infantile external genitalia | Underdeveloped labia; small uterus |
| Sparse pubic hair | Some pubic/axillary hair may develop (adrenarche preserved) |
| No growth spurt | Continued short stature |
| Gonadotropins | FSH and LH markedly elevated (hypergonadotropic hypogonadism) |
| Oestrogen | Very low (streak gonads) |
"Secondary sexual characteristics do not develop in 90% of affected females." - Langman's Medical Embryology
Adult rule: Short stature + primary amenorrhoea → Turner syndrome until proven otherwise. - Berek & Novak's Gynecology
6. COMPLETE CLINICAL FEATURE CATALOGUE
Head & Neck
- Low posterior hairline
- Webbed neck (pterygium colli) - bilateral
- Short, broad neck
- Low-set, malformed ears
- High-arched palate
- Micrognathia
- Triangular mouth
- Epicanthal folds (in some)
- Ptosis
- Strabismus/amblyopia
Chest / Cardiovascular
- Shield chest (broad, barrel-shaped)
- Widely spaced nipples
- Bicuspid aortic valve (BAV) - 30-50%
- Coarctation of the aorta - 30% (most common cause of childhood death in TS)
- Aortic root dilation - present in 30%
- 100-fold higher risk of aortic dissection
- ASD, VSD
- Hypertension (even without cardiac or renal cause)
- Aortic dissection risk in pregnancy - 2% or more → relative contraindication for pregnancy
Upper Limb
- Cubitus valgus (increased carrying angle)
- Short 4th (and 5th) metacarpal
- Lymphoedema of hands (neonatal period)
- Clinodactyly, hyperconvex nails
Renal
- Horseshoe kidney - most common renal anomaly
- Pelvic kidney
- Duplicated collecting system
- Increased risk of urinary tract infections
- Nephrocalcinosis
- Hypertension from renal anomalies
Skin
- Multiple melanocytic naevi (pigmented)
- Increased risk of melanoma
- Alopecia areata, halo nevi
- Keloid formation
- Cutis laxa, cutis hyperelastica
- Vitiligo
Hearing
- Recurrent otitis media (AOM + OME) in childhood
- Progressive mid-to-high frequency sensorineural hearing loss (SNHL) - appears in school age and early adulthood
- Cholesteatoma - not infrequent
- Combined conductive + sensorineural losses common
- Scott-Brown's Otorhinolaryngology
Endocrine / Metabolic
- Hypothyroidism (autoimmune - Hashimoto's) - up to 50% with isochromosome Xq
- Type 2 Diabetes Mellitus - impaired glucose tolerance
- Insulin resistance
- Dyslipidaemia
- Hypertension (independent of cardiac/renal causes)
- Osteoporosis (oestrogen deficiency from early age)
Gastrointestinal
- Inflammatory bowel disease (IBD) - increased incidence
- Celiac disease - higher prevalence
- Hepatic dysfunction / elevated liver enzymes
- Telangiectasias of the intestinal wall
Gonadal / Reproductive
- Streak ovaries - white fibrous bands; no follicles
- Primary amenorrhoea (90% of cases)
- Infertility - virtually universal in classic 45,X
- However: some mosaic patients (45,X/46,XX) have spontaneous fertility
- Uterus: present and functional (can carry donor egg pregnancy)
- Increased risk of gonadoblastoma if Y chromosome material present
Musculoskeletal
- Short stature (SHOX haploinsufficiency)
- Scoliosis, kyphosis
- Short 4th metacarpal
- Madelung deformity (radial curvature of the wrist)
- Increased joint laxity
- Reduced bone mineral density → osteoporosis
Neurocognitive
- Intelligence: usually normal overall IQ
- Specific deficits:
- Visuospatial processing ("space-form blindness") - inability to appreciate shape and spatial relationships
- Maths difficulties
- Non-verbal learning disability
- Attention deficit
- Verbal IQ typically preserved
- Social anxiety and difficulties common
7. PATHOLOGY - OVARIES
- Initially normal ovarian development in utero
- Post-birth: accelerated follicular atresia
- Result: streak gonads = white fibrous stroma completely devoid of primordial follicles
- Histologically: no oocytes; fibrotic connective tissue
- The uterus and vagina develop normally (Müllerian structures do not depend on ovarian function for initial development)
- Streak gonads with Y material → gonadoblastoma risk
8. ENDOCRINOLOGY
| Hormone | Finding |
|---|---|
| FSH | ↑↑↑ Markedly elevated (hypergonadotropic) |
| LH | ↑↑ Elevated |
| Oestradiol | ↓↓ Very low/undetectable |
| AMH (Anti-Müllerian Hormone) | ↓↓ Very low (reflects ovarian reserve) |
| GH axis | Normal GH but reduced IGF-1 in some |
| Thyroid | ↑ TSH if hypothyroid (autoimmune) |
| Glucose/insulin | Insulin resistance pattern |
Pattern = Hypergonadotropic Hypogonadism (primary ovarian failure with high gonadotropins)
9. DIAGNOSIS
Who to Investigate?
| Age | Presenting Feature |
|---|---|
| Prenatal | Cystic hygroma on US; nuchal translucency ↑; IUGR; fetal hydrops |
| Neonatal | Lymphoedema of hands/feet; cardiac defect |
| Infancy/childhood | Recurrent otitis media + short stature (ENT red flag) |
| School age | Short stature below 3rd centile; falling off growth curve |
| Puberty | Failure of breast development; primary amenorrhoea |
| Adult | Short stature + primary amenorrhoea (classic adult presentation) |
Shortest girl in her class with ear problems = Think Turner Syndrome. - Scott-Brown's Otorhinolaryngology
Diagnostic Tests
| Test | Finding / Role |
|---|---|
| Peripheral blood karyotype | Gold standard - 45,X; mosaic forms |
| FISH | Rapid detection of X/Y; detect Y chromosome material |
| Microarray / SNP array | More sensitive for mosaicism; structural X abnormalities |
| NIPT (prenatal) | Detects monosomy X; high sensitivity |
| Amniocentesis/CVS | Definitive prenatal karyotype |
Even in classic Turner stigmata: always karyotype to exclude Y chromosome material. If Y material found → gonadectomy recommended. - Berek & Novak's Gynecology
Additional investigations at diagnosis:
| System | Test |
|---|---|
| Cardiac | Echocardiogram (BAV, coarctation, aortic root); ECG; blood pressure both arms |
| Renal | Renal ultrasound (horseshoe kidney, duplicated system) |
| Thyroid | TSH, free T4, anti-TPO antibodies |
| Metabolic | Fasting glucose, HbA1c, lipid profile |
| Bone | DEXA scan (baseline) |
| Hearing | Audiogram |
| Eyes | Ophthalmology (strabismus, ptosis, colour blindness) |
| GI | Coeliac serology (anti-tTG IgA) |
| Hormones | FSH, LH, oestradiol, AMH |
10. MANAGEMENT - MULTIDISCIPLINARY & AGE-SPECIFIC
A. Neonatal / Infant
- Confirm karyotype (blood lymphocytes) - urgent if lymphoedema of hands/feet present
- Echocardiogram - exclude coarctation of aorta (life-threatening); bicuspid aortic valve
- Measure blood pressure in all 4 limbs (upper > lower by >10 mmHg = coarctation)
- Renal ultrasound - horseshoe kidney, structural anomaly
- Genetic counselling for parents
- Multidisciplinary referral: paediatric cardiologist, endocrinologist, urologist, ENT
- No hormonal treatment needed at this age
- Lymphoedema management: gentle massage, compression stockings if severe
B. Infancy / Early Childhood
- Growth monitoring on Turner-specific growth charts
- ENT surveillance - hearing tests, tympanometry; prompt insertion of ventilation tubes (grommets) for OME with hearing loss - do NOT delay
- Audiological assessment annually
- Monitor thyroid function (TSH) - autoimmune thyroid disease from early childhood
- Developmental assessment - speech, language, motor
C. School Age - Starting Growth Hormone
Growth Hormone (GH) Therapy:
- Indication: Short stature in Turner syndrome
- Mechanism: GH works via SHOX upregulation + IGF-1 stimulation; Turner-specific higher doses needed
- When to start: As early as 2-5 years when height falls below 5th centile for age
- Dose: Higher than in GH deficiency: ~0.375 mg/kg/week (~50 µg/kg/day)
- Goal: Increase adult height; without treatment, final height rarely exceeds 150 cm in 45,X
- With GH treatment: Can add 5-8 cm to adult height (evidence from multiple RCTs)
- Side effects: Common GH side effects; watch for scoliosis, slipped capital femoral epiphysis, raised intracranial pressure
- Monitoring: Growth velocity, IGF-1, bone age, glucose
- Continue until growth velocity slows and bone age > 14 years (or adult height achieved)
- Harrison's 22e; Andrews' Diseases of the Skin
Oxandrolone (anabolic steroid):
- Sometimes added to GH in girls >8 years to further enhance height
- Dose: 0.03-0.05 mg/kg/day
- Monitor for virilisation (voice changes, clitoral enlargement)
D. Puberty - Oestrogen Replacement Therapy
Why: Streak ovaries produce no oestrogen → no puberty will occur spontaneously in ~90%
Goals of oestrogen therapy:
- Induce breast development (thelarche)
- Uterine development (for potential future pregnancy)
- Support height growth
- Prevent osteoporosis
- Cardiovascular health
- Psychological well-being
Protocol:
| Phase | Oestrogen | Dose | Duration |
|---|---|---|---|
| Initiation | Natural 17β-oestradiol (transdermal patch preferred) | Ultra-low dose (0.5-1 µg/day transdermal; 0.25 mg/day oral) | ~2-3 years |
| Dose escalation | Increase gradually | Over 2-4 years to adult replacement dose | Mimics normal pubertal progression |
| Adult dose | Full replacement | Adult dose | Lifelong |
| Add progestogen | Only once breakthrough bleeding occurs OR after 2 years | Cyclic (12-14 days/month) | To induce withdrawal bleeds + protect endometrium |
"Most physicians now initiate low-dose oestrogen therapy to induce puberty at an age-appropriate time (~11 years)." - Harrison's 22e
Route: Transdermal oestradiol preferred over oral (avoids first-pass hepatic effects; better bone and cardiovascular outcomes; better IGF-1 response)
E. Cardiovascular - Lifelong
- Echo + MRI aorta at diagnosis and every 5 years (annually if aortic root ≥3 cm)
- Coarctation: surgical repair or balloon dilatation
- Bicuspid aortic valve: monitor for stenosis and regurgitation
- Aortic dissection risk: 100-fold higher than general population
- Highest risk triggers: pregnancy, hypertension, exercise
- Aortic root >3.5 cm → counselled strongly against pregnancy
- BP monitoring - treat hypertension aggressively
- Antibiotic prophylaxis before dental/surgical procedures (if BAV)
- Cardiology follow-up: lifelong
F. Fertility & Reproductive Health
Current fertility options:
| Option | Details |
|---|---|
| Donor egg IVF + embryo transfer | Most successful; uterus is normal and functional |
| Spontaneous conception | Very small percentage with mosaic TS (45,X/46,XX) |
| Ovarian tissue cryopreservation | Experimental; possible in young girls with residual follicles |
| Fertility preservation | Consider in adolescents with mosaicism and some ovarian function |
Pregnancy risks in TS:
- Aortic dissection risk 2% or more during pregnancy
- Hypertension common in pregnancy
- Pre-eclampsia
- American Society for Reproductive Medicine (2012): TS is a relative contraindication for pregnancy
- If pursuing pregnancy: pre-pregnancy cardiac MRI + aortic root measurement; monthly cardiac surveillance throughout pregnancy; deliver in tertiary centre
Y chromosome material:
- 5-10% of TS patients have Y chromosome sequences (complete Y = 45,X/46,XY; or Y fragments)
- Risk: gonadoblastoma (benign but precursor to malignancy - dysgerminoma, teratoma, embryonal carcinoma, endodermal sinus tumour)
- Management: prophylactic laparoscopic gonadectomy at time of diagnosis
- If no tumour dissemination: uterus left in situ for donor IVF
- Increasingly: individualised decision incorporating potential gonadal function and patient preference
- Berek & Novak's Gynecology; Harrison's 22e
G. Bone Health
- Oestrogen replacement critical (prevent osteoporosis)
- DEXA scan at baseline, then every 2-5 years
- Calcium and vitamin D supplementation from childhood
- Weight-bearing exercise encouraged
- Monitor for scoliosis with GH therapy
H. ENT / Audiology
- Annual audiogram from diagnosis
- Grommets (ventilation tubes) for OME with hearing loss - early intervention
- Monitor for cholesteatoma
- Progressive SNHL: hearing aids when indicated
- Speech therapy if language delay
I. Autoimmune / Endocrine Surveillance
- Thyroid function (TSH, anti-TPO): annually from age 4 years
- Fasting glucose / HbA1c: annually from adolescence
- Lipid profile: from adolescence
- Coeliac disease screen (anti-tTG IgA): every 2-5 years
- Liver function tests: annually in adulthood (elevated liver enzymes common)
J. Ophthalmology
- Strabismus, ptosis, amblyopia - treat early (critical periods for vision)
- Colour blindness more common in X-monosomic patients
K. Neurodevelopmental / Educational
- Neuropsychological testing for visuospatial learning disabilities
- Educational psychology assessment
- Targeted maths/spatial skills intervention
- Screen for ADHD features
- Psychological support - anxiety is very common
- Peer support groups (invaluable)
11. LONG-TERM SURVEILLANCE - SUMMARY TABLE
| Domain | Frequency | Action |
|---|---|---|
| Growth | Every 3-6 months | GH therapy; Turner growth charts |
| Cardiovascular | Annually (echo/MRI) | BP, aortic dimensions, BAV surveillance |
| Thyroid | Annually | TSH, free T4, anti-TPO |
| Glucose/HbA1c | Annually | DM screening |
| Lipids | Annually | Dyslipidaemia |
| Liver enzymes | Annually | Hepatic dysfunction |
| Bone density (DEXA) | Every 2-5 years | Osteoporosis prevention |
| Hearing | Annually | Audiogram; hearing aids |
| Renal | At diagnosis; then PRN | UTIs, hypertension |
| Celiac screen | Every 2-5 years | Anti-tTG IgA |
| Skin / nevi | Annually | Melanoma surveillance |
| Mental health | Ongoing | Anxiety, depression |
| Oestrogen/hormone levels | Every 6-12 months | Adequacy of HRT |
| Fertility counselling | From adolescence | Donor egg options |
"Long-term follow-up includes careful surveillance of sex hormone replacement and reproductive function, bone mineralisation, cardiac function and aortic root dimensions, blood pressure, weight and glucose tolerance, hepatic and lipid profiles, thyroid function, celiac disease screening, skin examination, and hearing. This service is provided by a dedicated Turner syndrome clinic in some centres." - Harrison's 22e
12. DIFFERENTIALS
| Condition | Key Distinguishing Features |
|---|---|
| Noonan syndrome | Normal karyotype (46,XX or 46,XY); autosomal dominant; similar phenotype (webbed neck, short stature, CHD - pulmonary stenosis rather than coarctation); males affected |
| Multiple pterygium syndrome (Escobar) | Autosomal recessive; joint contractures; mimics TS |
| GH deficiency | No dysmorphic features; bone age delayed; low IGF-1; karyotype normal |
| Constitutional delay | No dysmorphic features; normal karyotype; family history |
| Primary ovarian insufficiency | Normal karyotype; may have autoimmune cause; no somatic features |
| Hypothyroidism | Short stature; goitre; normal karyotype |
13. QUICK EXAM PEARLS
- TS = 45,X → only viable monosomy; 98% abort spontaneously
- Most common chromosomal disorder in humans (vast majority miscarry)
- Missing X is usually paternal (75%) - father's sperm failed to contribute sex chromosome
- Neonatal sign: Lymphoedema of dorsum of hands AND feet → KARYOTYPE IMMEDIATELY
- Cardiac: Coarctation of aorta + bicuspid aortic valve → most common cause of death in childhood
- Aortic dissection risk: 100-fold increased; 2% risk in pregnancy → relative contraindication for pregnancy
- SHOX gene on Xp22 → haploinsufficiency → short stature + skeletal defects
- Ovaries: initially normal, then accelerated atresia → streak gonads (fibrotic, no follicles)
- GH therapy started early (age 2-5 years): can add ~5-8 cm to final height
- Oestrogen therapy started at ~11 years (age-appropriate puberty induction): SLOWLY escalate over 2-4 years
- Add progestin after breakthrough bleeding or 2 years of oestrogen
- Y chromosome material (5-10% of TS) → gonadoblastoma risk → prophylactic gonadectomy
- Fertility: Donor egg IVF possible (uterus normal); AMSRM considers TS a relative contraindication for pregnancy due to cardiac risk
- Hearing: Recurrent OME in childhood → progressive SNHL in adulthood → annual audiogram mandatory
- Hypothyroidism in 50% with isochromosome Xq → annual thyroid screening
- Intellect: Normal overall IQ but specific visuospatial ("space-form blindness") and maths deficits
- Mosaic TS (45,X/46,XX): milder phenotype; rare spontaneous pregnancy possible
- Presentation triads to memorise:
- Neonatal: Lymphoedema + neck webbing + cardiac defect
- Childhood: Short stature + recurrent ear infections + no puberty
- Adult: Short stature + primary amenorrhoea + streak ovaries
Sources:
- Robbins & Kumar Basic Pathology
- Robbins, Cotran & Kumar Pathologic Basis of Disease
- Berek & Novak's Gynecology
- Harrison's Principles of Internal Medicine 22e (2025)
- Langman's Medical Embryology
- Andrews' Diseases of the Skin
- Scott-Brown's Otorhinolaryngology Head & Neck Surgery
- Campbell-Walsh-Wein Urology
- Recent evidence: Khan N et al., Turner Syndrome: where are we? Orphanet J Rare Dis 2024 (PMID 39198906) | Turner HE et al., Approach to the Patient with Turner Syndrome, JCEM 2025 (PMID 40971503) | Wan KL et al., Turner Syndrome, J Paediatr Child Health 2025 (PMID 40642882) | Society for Endocrinology Guideline on Female Hypogonadism 2024 (PMID 39031660)
Teache me as neonatatologist, paeditrcian in depth topic- Down's syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Down syndrome" AND management AND pediatric
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
I now have comprehensive content from multiple authoritative sources. Here is the full in-depth teaching:
Down Syndrome (Trisomy 21) - In-Depth Teaching for Neonatologists & Paediatricians
1. DEFINITION & HISTORY
Down syndrome is a chromosomal disorder caused by the presence of an extra copy of chromosome 21 (trisomy 21), resulting in characteristic facial features, intellectual disability, hypotonia, and multi-system congenital anomalies.
First described in 1866 by Langdon Down. The chromosomal basis (trisomy 21) was established in 1959.
- Adams and Victor's Principles of Neurology
It is the most common chromosomal disorder and the most common genetic cause of intellectual disability.
2. EPIDEMIOLOGY
| Parameter | Data |
|---|---|
| Incidence | ~1 in 600-700 live births |
| Most common chromosomal anomaly | Yes - most common in every large series of intellectual disability |
| Accounts for | ~10% of all cases of severe intellectual disability |
| Maternal age <20 years | 1 in 1,550 live births |
| Maternal age 35 years | 1 in 300 live births |
| Maternal age 40 years | 1 in 100 live births |
| Maternal age >45 years | 1 in 25 live births |
Maternal age has a strong influence on incidence. In 95% of cases, the extra chromosome is of maternal origin (nondisjunction in the ovum). No effect of paternal age found.
- Robbins & Kumar Basic Pathology
3. GENETICS - KARYOTYPES & MECHANISMS
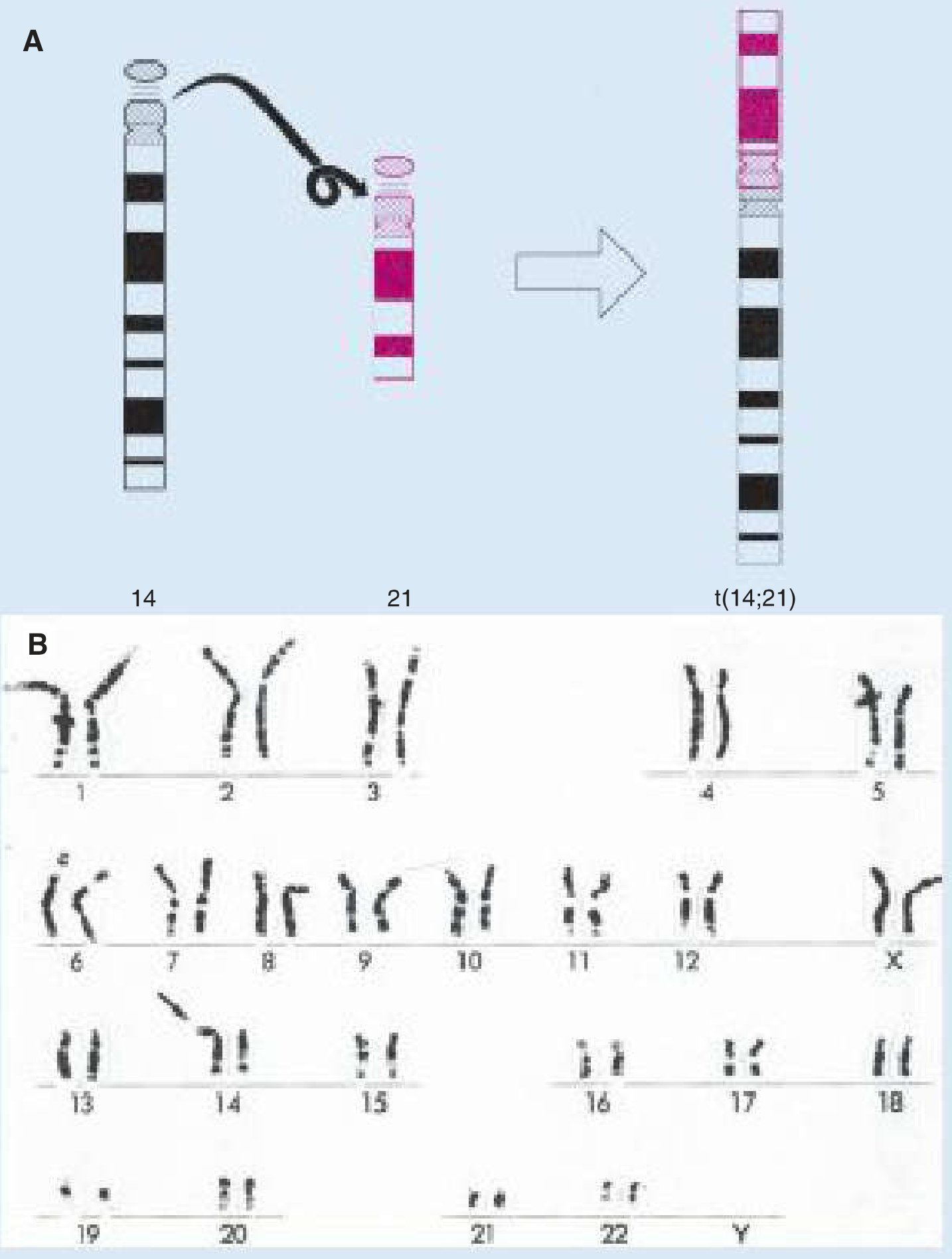
Langman's Medical Embryology - Fig 2.7: A. Translocation t(14;21) - carrier parent is clinically normal but at risk of Down syndrome offspring. B. Full karyotype of translocation Down syndrome.
Three Types - Master This Table
| Type | Frequency | Karyotype | Mechanism | Parental Carrier? | Recurrence Risk |
|---|---|---|---|---|---|
| Trisomy 21 (Free trisomy) | ~95% | 47,XX(or XY),+21 | Meiotic nondisjunction; 75% in oogenesis | No (parents normal) | Low (~1%); increases with maternal age |
| Translocation | ~4% | 46,XX,der(14;21)(q10;q10),+21 | Long arm of chromosome 21 translocated onto chr 14 (or 13, 15, 22) - Robertsonian translocation | YES - one parent is carrier (balanced translocation); 46 chromosomes but carrier is normal | HIGH - up to 10-15% if mother is carrier; 2-5% if father |
| Mosaicism | ~1% | 46,XX/47,XX,+21 | Mitotic nondisjunction after fertilisation; some cells 46, some 47 | No | Low |
Critical Points on Translocation Type
- Translocation parent has 46 chromosomes (balanced) - phenotypically NORMAL
- But they carry the translocated chromosome → offspring risk
- If mother is carrier: ~10-15% risk of Down syndrome offspring
- If father is carrier: ~2-5% risk
- Familial Down syndrome: Always investigate the translocation type
- The two genes of greatest interest on chromosome 21: DYRK1A and DSCR1 - involved in neurodevelopment
- Adams and Victor; Robbins Pathology
Mosaic Down Syndrome
- Variable phenotype depending on proportion of trisomic cells
- Milder features and intelligence may be average or near-average
- Some mosaics are of normal intelligence
4. PATHOGENESIS - WHY DO THESE FEATURES ARISE?
Gene Dosage Effect
- An extra copy of chromosome 21 means ~300 extra genes expressed at ~150% of normal
- Key genes:
- COL6A1, COL6A2 (type VI collagen) → generalised joint laxity + orthopaedic problems
- APP (amyloid precursor protein) → Alzheimer disease (chromosome 21 location)
- DYRK1A → intellectual disability, neurodevelopment
- SOD1 (superoxide dismutase) → oxidative stress → premature ageing
- ERG/ETS2 → leukaemia predisposition
Intellectual Disability Mechanism
- Generalised cerebral maldevelopment
- Hypoplasia of the frontal lobes and cerebellum
- Reduced number of neurones and simplified dendritic branching
- Lower synaptogenesis
5. CLINICAL FEATURES - COMPLETE CATALOGUE
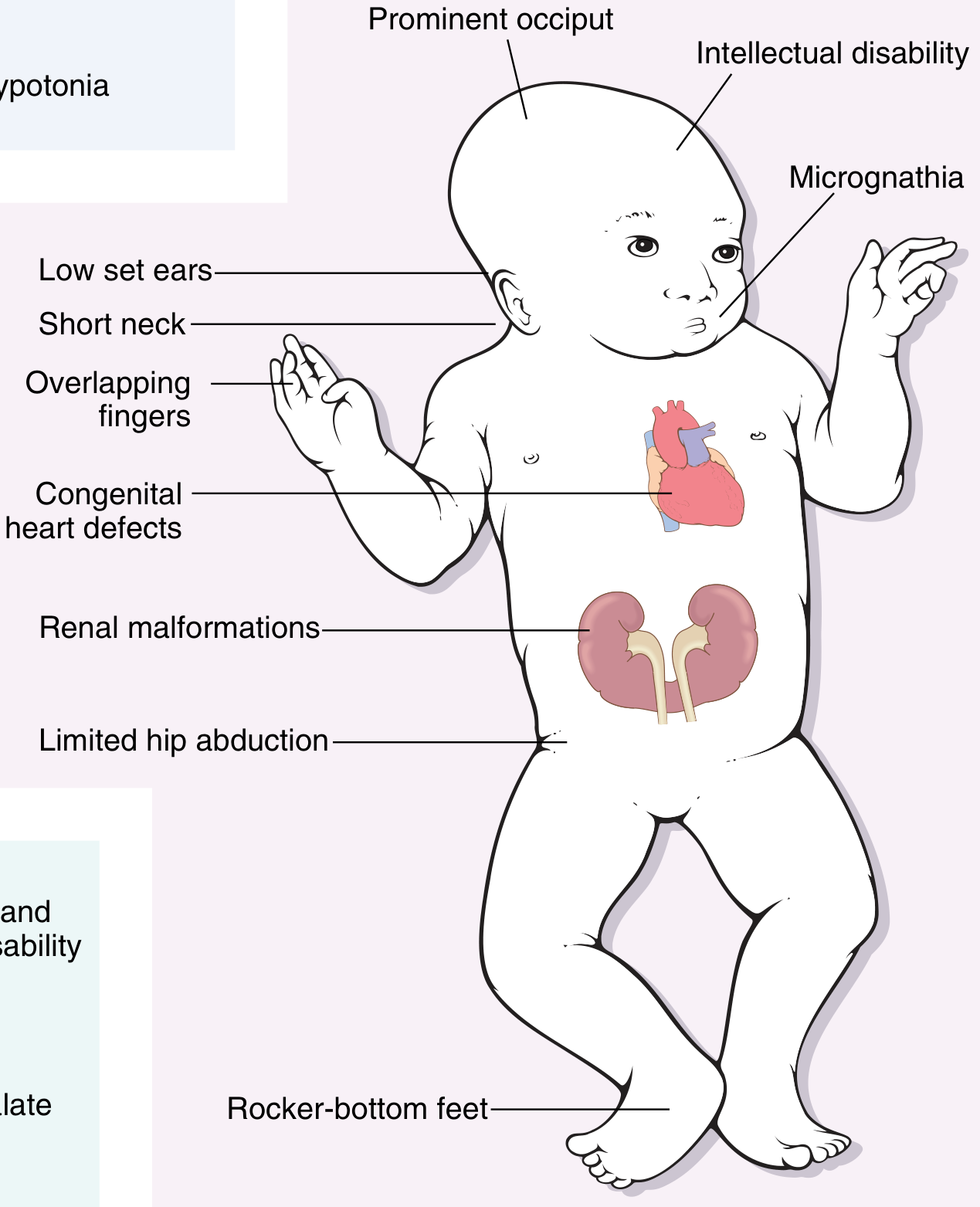
Robbins & Kumar Basic Pathology - Clinical features summary: Hypotonia, intellectual disability, congenital heart defects, renal malformations.
A. Head & Face (Characteristic - Allow Clinical Diagnosis at Birth)
| Feature | Description |
|---|---|
| Flat facial profile | Flat face overall; hypoplastic maxillae |
| Brachycephaly | Round head; short anteroposterior diameter |
| Flat occiput | Posterior flattening of skull |
| Upward-slanting palpebral fissures | Eyes slant upward and outward (oblique) |
| Epicanthic folds | Extra skin fold at medial canthi (covering inner angle of eye) |
| Brushfield spots | Grey-white specks of depigmentation in the irides |
| Flat nasal bridge | Wide, poorly developed nasal bridge |
| Small mouth / high-arched palate | Relative macroglossia |
| Large, fissured, protruding tongue | Heavily grooved; protrudes (relative to small oral cavity) |
| Ears | Small, low-set, oval with small lobes |
| Short neck | Excess skin at posterior neck |
| Low posterior hairline |
B. Neurological / Developmental
| Feature | Description |
|---|---|
| Hypotonia | Profound; most striking neonatal finding; reduced/absent Moro reflex; feeding difficulties |
| Intellectual disability | Present in ALL patients; variable degree; IQ range 20-70 (median 40-50) |
| Delayed motor milestones | Most don't walk until 3-4 years |
| Delayed speech | >90% talk by age 5 years |
| Personality | Placid, docile, affectionate - characteristic |
| Seizures | Increased incidence (infantile spasms in infancy; focal seizures later) |
C. Hands & Limbs
| Feature | Description |
|---|---|
| Single palmar crease (Simian crease) | Single transverse crease replacing normal 2 creases - pathognomonic |
| Clinodactyly | 5th finger curved (short, hypoplastic middle phalanx) |
| Broad, stubby hands | Short digits |
| Sandal gap | Wide gap between 1st and 2nd toes |
| Hyperextensible joints | Generalised ligamentous laxity (COL6A1/COL6A2) |
| Short stature | Adult height rarely exceeds that of a 10-year-old child |
D. Cardiovascular (40% of patients)
- Most common cause of death in infancy and early childhood
- Endocardial cushion defects are most characteristic:
- Atrioventricular septal defect (AVSD) / Endocardial cushion defect - most characteristic
- Atrial septal defect (ASD)
- Ventricular septal defect (VSD)
- Patent ductus arteriosus (PDA)
- Tetralogy of Fallot (less common)
E. Gastrointestinal
| Condition | Frequency | Notes |
|---|---|---|
| Duodenal atresia | ~12% | "Double bubble" on AXR; "apple-peel" appearance |
| Hirschsprung disease | 2-3% | Neonatal intestinal obstruction; aganglionic colon |
| Oesophageal atresia | Increased | Associated with other GI atresias |
| Imperforate anus | Increased | |
| Coeliac disease | Increased | Screen regularly |
Down syndrome + Hirschsprung disease = important association for paediatric exams. 10% of Hirschsprung patients have Down syndrome. - Goldman-Cecil Medicine
F. Ophthalmological
- Cataracts (congenital or developing in childhood) - screen at birth
- Refractive errors (myopia, hyperopia)
- Strabismus and nystagmus
- Brushfield spots (iris speckling)
- Blepharitis
- Keratoconus (in older patients)
G. ENT / Hearing
- Conductive hearing loss - very common; due to recurrent otitis media + structural middle ear abnormalities
- Sensorineural hearing loss - also occurs
- Recurrent AOM and OME - major problem; Eustachian tube dysfunction
- Subglottic stenosis - important for anaesthesia
- Absent/decreased frontal and sphenoid sinuses (hypopneumatisation)
- KJ Lee's Essential Otolaryngology
H. Endocrine
- Hypothyroidism - occurs in 15-20%; can be congenital or acquired (Hashimoto's)
- Diabetes mellitus (both type 1 and type 2)
- Abnormal thyroid autoantibodies common
I. Immunological
- Abnormal immune responses - particularly T-cell function defects
- Increased susceptibility to serious infections, especially pneumonia
- Increased autoimmune diseases: thyroiditis, coeliac disease, alopecia areata, vitiligo
- Robbins Pathology - Down syndrome and autoimmune disease review (PMID 38913142, 2024)
J. Haematological / Oncological
- 10-20 fold increased risk of leukaemia (both ALL and AML)
- Transient Myeloproliferative Disease (TMD) / Transient Abnormal Myelopoiesis (TAM):
- Occurs in 5-10% of neonates with Down syndrome
- Spontaneously resolves in most within 3 months
- However, ~20-30% of TAM cases develop AMKL (acute megakaryoblastic leukaemia) by age 4
- Screening: CBC in all neonates with Down syndrome
- JAMA Oncology review (PMID 37440251, 2023); Haematologica review (PMID 37439336, 2023)
K. Musculoskeletal / Orthopaedic
- Generalised ligamentous laxity (due to abnormal type VI collagen - COL6A1, COL6A2 on chromosome 21)
- Atlantoaxial instability (AAI):
- Present in 10-20% of Down syndrome patients
- Atlanto-dens interval (ADI): normal <4 mm; concern >4-5 mm; surgery if >10 mm or neurological symptoms
- ~30% with AAI develop premature cervical spondylosis
- ~1-2% develop symptomatic spinal cord dysfunction
- Screening X-ray at age 3 years (AAP guideline); sooner if symptomatic
- Contact sports/gymnastics/diving restricted based on ADI
- Pre-anaesthetic cervical spine assessment mandatory (risk of cord injury during intubation)
- Scoliosis - 50% of cases
- Flat feet (pes planus) + metatarsus primus varus
- Hip instability
- Patellar dislocation
- Slipped capital femoral epiphysis (SCFE)
- Miller's Review of Orthopaedics
L. Neurological Complications
- Alzheimer Disease - most significant long-term complication:
- APP gene (amyloid precursor protein) is on chromosome 21
- Extra copy → overproduction of amyloid → β-amyloid plaques and tau tangles
- Virtually ALL patients with trisomy 21 older than 40 develop Alzheimer's histologically
- 30% develop dementia by age 60; 50% by age 70
- Early signs in DS patients: inattentiveness, reduced speech, loss of skills, withdrawal, behaviour change, seizures (NOT the classic memory loss presentation)
- Goldman-Cecil Medicine; Adams and Victor's Neurology
- Moyamoya disease - progressive carotid vasculopathy → stroke risk
- Epilepsy - infantile spasms in infancy; other seizure types
- Stroke - embolic (from cardiac defects); Moyamoya; cerebral venous sinus thrombosis
M. Renal / Urological
- Renal malformations (horseshoe kidney, hydronephrosis)
- Cryptorchidism and hypospadias
- Urinary tract infections
N. Sleep
- Obstructive sleep apnoea (OSA) - very common due to:
- Midface hypoplasia
- Macroglossia
- Hypotonia of pharyngeal muscles
- Adenotonsillar hypertrophy
- Smaller airway diameter
- Screen all DS children with sleep study
- Adenotonsillectomy often required
6. PRENATAL SCREENING & DIAGNOSIS
First Trimester Screening (10-14 weeks)
| Test | Finding in Down Syndrome | Sensitivity |
|---|---|---|
| Nuchal translucency (NT) ultrasound | ↑↑ Increased (>3 mm at 11-14 weeks) | 75-80% alone; 90%+ combined |
| Maternal serum β-hCG | ↑ Increased | - |
| PAPP-A (pregnancy-associated plasma protein A) | ↓ Decreased (poor placental function) | - |
| Combined first-trimester test (NT + β-hCG + PAPP-A) | Combined risk calculation | ~90% sensitivity at 5% FPR |
Second Trimester Screening
| Test | Finding in Down Syndrome |
|---|---|
| Maternal serum AFP | ↓ Decreased |
| β-hCG | ↑ Increased |
| Unconjugated oestriol (uE3) | ↓ Decreased |
| Inhibin A | ↑ Increased |
| Triple test (AFP + β-hCG + uE3) | Detects ~70% at 5% FPR |
| Quadruple test (+inhibin A) | Detects ~80% at 5% FPR |
Second trimester US findings ("soft markers"):
- Nuchal fold thickening (≥6 mm at 15-21 weeks)
- Short femur/humerus
- Echogenic bowel
- Pyelectasis
- Echogenic intracardiac focus (EIF)
- Choroid plexus cysts (more specific for Trisomy 18)
- Absent/hypoplastic nasal bone
Cell-Free DNA (cfDNA) / NIPT (Non-Invasive Prenatal Testing)
- ~5-10% of total cell-free DNA in maternal blood is fetal
- Next-generation sequencing of chromosome 21-linked genes
- Sensitivity >99% for Trisomy 21; specificity >99%
- Superior to traditional screening; increasingly first-line
- All positive NIPT results must be confirmed by conventional cytogenetics (amniocentesis)
- Robbins & Kumar Basic Pathology
Definitive Prenatal Diagnosis
| Procedure | Timing | Risk |
|---|---|---|
| Chorionic villus sampling (CVS) | 10-13 weeks | ~0.5-1% miscarriage |
| Amniocentesis | 15-20 weeks | ~0.3-0.5% miscarriage |
7. NEONATAL ASSESSMENT - IMMEDIATE MANAGEMENT
Diagnosis at Birth
- Clinical recognition - characteristic features permit recognition at birth
- Confirm by karyotype (blood sample for cytogenetics) - mandatory even if clinically obvious
- Determines whether trisomy 21, translocation, or mosaic → critical for recurrence counselling
- If translocation type → parental karyotype mandatory
Neonatal Investigations (AAP Guidelines) - Harriet Lane Handbook
| Investigation | Rationale |
|---|---|
| Echocardiogram | 40% have CHD; may be silent; AVSD most common; diagnose before symptoms |
| CBC with differential | Screen for transient myeloproliferative disease (TMD) |
| Thyroid function (TSH) | Hypothyroidism (congenital) |
| Hearing screen (AABR) | Conductive and sensorineural hearing loss |
| Ophthalmology referral | Cataracts (screen at birth); nystagmus, strabismus |
| Feeding assessment | Hypotonia + macroglossia → feeding difficulties |
| Karyotype | Confirm type; recurrence risk counselling |
| Renal ultrasound | Renal malformations |
| Abdominal examination | Hirschsprung disease; duodenal atresia (if vomiting bilious) |
| Early intervention referral | Physiotherapy, occupational therapy, speech therapy |
Specific Neonatal Concerns
Feeding:
- Profound hypotonia → poor suck and swallow
- Nasogastric tube feeding may be required initially
- Breastfeeding encouraged but often requires lactation support
- Monitor weight carefully
Cardiovascular:
- Echocardiogram before discharge or within first week
- AVSD may not be clinically apparent in first days (pulmonary vascular resistance still high)
- Refer to paediatric cardiology
Transient Myeloproliferative Disease (TMD):
- 5-10% of neonates with DS
- Peripheral blood smear: blast cells (megakaryoblasts)
- Most resolve spontaneously within 3 months
- Monitor CBC closely
- 20-30% risk of developing AMKL (leukaemia) by age 4 → surveillance required
Atlantoaxial Instability:
- Cannot screen at birth (physiological laxity)
- BUT: cervical spine X-ray required before any anaesthesia to assess ADI
- Cervical spine screening X-ray at age 3 years (or sooner if symptomatic)
8. DEVELOPMENTAL FEATURES & MILESTONES
| Milestone | Typical DS Child | Normal Range |
|---|---|---|
| Walking | 3-4 years | 12-15 months |
| First words | Delayed; most talk by 5 years | 12 months |
| Toilet training | 3-7 years | 2-3 years |
| Reading | Variable; some achieve functional literacy | - |
| IQ range | 20-70 (median 40-50) | 85-115 |
Over 90% of children with Down syndrome talk by age 5. With appropriate intervention, most can attend school and manage some activities of daily living. - Goldman-Cecil Medicine
9. MANAGEMENT - AGE-SPECIFIC & MULTIDISCIPLINARY
A. Neonatal (0-1 month)
- Confirm diagnosis (karyotype) + parental counselling (genetic counselling)
- Echocardiogram
- CBC (TMD screen)
- Thyroid function
- Hearing screen
- Ophthalmology
- Early referral to early intervention programme
- Parent support groups
B. Infancy (1-12 months)
- Physiotherapy - hypotonia management, motor development
- Speech-language therapy - early communication, feeding
- Occupational therapy - fine motor
- Cardiac surgery if CHD (typically within first 3-6 months for AVSD - before pulmonary hypertension develops)
- Annual thyroid function tests (TSH) - from first year
- Annual CBC (TMD → AMKL surveillance)
- Hearing assessment (ABR, tympanometry) - at 6 months and annually
- Vision - ophthalmology review
C. Childhood
- Full health supervision as per AAP Down Syndrome Health Supervision Guidelines (current version)
- Growth charts - use Down syndrome-specific growth charts
- Thyroid function: Annually
- CBC: Annually (leukaemia surveillance)
- Hearing: Annually
- Vision: Every 2 years
- Coeliac disease screen (anti-tTG IgA): at diagnosis and periodically
- Cervical spine X-ray at age 3 years (atlantoaxial instability screening):
- ADI <4.5 mm: no restrictions
- ADI 4.5-10 mm: avoid contact sports, diving, gymnastics
- ADI >10 mm or neurological symptoms: C1-C2 fusion + neurosurgical referral
- Obstructive sleep apnoea: Overnight polysomnography; adenotonsillectomy if indicated
- Educational support: Inclusion in mainstream school with support; individual education plans (IEP)
- Behavioural support: ADHD features common; anxiety
- Dental: Regular review (periodontal disease common)
D. Adolescence
- Monitoring for hypothyroidism (annual TSH), diabetes, leukaemia
- OSA surveillance (worsens during adolescence)
- Transition from paediatric to adult care planning
- Atlantoaxial instability: Review cervical spine before any sport participation (Special Olympics requires cervical spine X-ray for selected sports)
- Mental health: Depression, anxiety, autistic spectrum features common
- Sexuality education; fertility (women with DS can have children; 50% risk of DS in offspring; men are usually infertile)
E. Adulthood - Alzheimer Disease Surveillance
- Baseline cognitive assessment at age 30-35 (before expected decline)
- Annual cognitive monitoring from age 40
- Early Alzheimer signs in DS: behaviour change, withdrawal, loss of skills, seizures (NOT classic memory loss)
- Cholinesterase inhibitors (donepezil) - some evidence of benefit; not universally recommended
- Antiepileptics for seizures
10. CARDIAC MANAGEMENT (Special Section)
| Defect | Features | Timing of Repair |
|---|---|---|
| AVSD (Endocardial cushion defect) | Most characteristic; large L-R shunt; pulmonary hypertension risk | 3-6 months (before irreversible Eisenmenger) |
| VSD | Common; variable size | 3-6 months if large |
| ASD | May close spontaneously | 2-5 years if persistent |
| PDA | Medical closure (indomethacin/ibuprofen); surgical if persistent | Early |
| Tetralogy of Fallot | Less common in DS; surgical repair | 3-6 months |
Pulmonary vascular disease (Eisenmenger syndrome) develops earlier in DS patients with large shunts than in normal children - repair must not be delayed.
11. LEUKAEMIA IN DOWN SYNDROME
| Type | Age | DS-Specific Feature |
|---|---|---|
| Transient Myeloproliferative Disease (TMD/TAM) | Neonatal | GATA1 somatic mutation; mostly self-resolving; 20-30% progress to AMKL |
| AMKL (Acute Megakaryoblastic Leukaemia) | 1-4 years | 500-fold higher risk in DS; responds well to treatment |
| ALL (Acute Lymphoblastic Leukaemia) | Childhood | 10-20x increased risk vs general population; good prognosis with modified chemotherapy |
| AML | Any age | Distinct biology in DS; more chemosensitive; dose reduction required (increased toxicity) |
DS-associated leukaemias are generally more chemosensitive than sporadic leukaemias. However, DS children have increased toxicity from standard chemotherapy doses and require protocol modification. - JAMA Oncology 2023 (PMID 37440251)
12. PRENATAL COUNSELLING & RECURRENCE RISK
| Type | Recurrence Risk |
|---|---|
| Trisomy 21 (nondisjunction) | Low - ~1% + maternal age risk; slightly higher than age-matched risk |
| Translocation (maternal carrier) | ~10-15% per pregnancy |
| Translocation (paternal carrier) | ~2-5% per pregnancy |
| Translocation 21;21 (Robertsonian) | 100% risk - all viable pregnancies will have DS |
| Mosaic parent | Variable; depends on gonadal mosaicism |
Parental karyotyping is mandatory in all translocation cases. If a parent has Robertsonian translocation 21;21, the recurrence risk is effectively 100% for all pregnancies.
13. PROGNOSIS & LIFE EXPECTANCY
- Improved dramatically with modern medical and surgical care
- Current life expectancy: ~55-60 years (some reaching 70s)
- Previously (pre-1980s): ~25 years
- Main causes of early mortality: CHD (infancy), infections
- Main cause of late mortality: Alzheimer disease (from 4th-5th decade)
- Quality of life: Most patients lead enjoyable, meaningful lives with appropriate support
- Many are semi-independent; some hold employment; meaningful social relationships
14. DIFFERENTIAL DIAGNOSIS
| Condition | Key Distinguishing Features |
|---|---|
| Zellweger syndrome | Hypotonia + dysmorphism but normal karyotype; peroxisomal disorder |
| Congenital hypothyroidism | Hypotonia + macroglossia + facial puffiness; normal karyotype; TSH elevated |
| Beckwith-Wiedemann | Macroglossia + macrosomia + hypoglycaemia; normal karyotype |
| Prader-Willi | Hypotonia + feeding difficulty; chromosome 15q deletion; will gain weight |
| Noonan syndrome | Webbed neck + cardiac defects; normal karyotype; PTPN11 mutation |
15. QUICK EXAM PEARLS
- Down syndrome = extra chromosome 21 → trisomy (95%), translocation (4%), mosaic (1%)
- Most common chromosomal disorder; most common genetic cause of intellectual disability
- 95% have extra chromosome of maternal origin (nondisjunction in oogenesis)
- Translocation type = FAMILIAL → always karyotype parents; recurrence risk up to 100% if t(21;21)
- At birth: Hypotonia is the most striking and universal neonatal finding
- Single palmar crease (Simian crease) + clinodactyly of 5th finger
- Cardiac defect in 40% → most common cause of death in childhood → AVSD most characteristic
- Echocardiogram, CBC, TFT, hearing screen - mandatory at birth
- Transient myeloproliferative disease (TMD) in 5-10% neonates → self-limiting but 20-30% → AMKL
- 10-20x risk of leukaemia (both ALL and AMKL)
- Atlantoaxial instability in 10-20% → cervical spine X-ray at age 3; mandatory pre-anaesthesia
- Virtually ALL DS patients >40 develop Alzheimer histologically → APP gene on chromosome 21
- Alzheimer in DS presents as: behaviour change, withdrawal, loss of skills (NOT classic memory loss)
- Hypothyroidism in 15-20% → annual TSH screening
- OSA very common → overnight polysomnography; adenotonsillectomy
- Duodenal atresia + DS → "Double bubble" on AXR → surgical emergency
- Hirschsprung disease - 10% of Hirschsprung patients have Down syndrome
- Subglottic stenosis → important airway consideration (anaesthesia/intubation)
- Life expectancy now ~55-60 years (was 25 years before modern care)
Sources:
- Robbins & Kumar Basic Pathology
- Robbins, Cotran & Kumar Pathologic Basis of Disease
- Adams and Victor's Principles of Neurology 12th Ed.
- Langman's Medical Embryology
- Goldman-Cecil Medicine
- Miller's Review of Orthopaedics 9th Ed.
- KJ Lee's Essential Otolaryngology
- Harriet Lane Handbook 23e (Johns Hopkins Hospital)
- Recent evidence: Down syndrome and leukaemia review Haematologica 2023 (PMID 37439336) | DS-associated leukaemia management, JAMA Oncol 2023 (PMID 37440251) | DS and autoimmune disease review, Clin Rev Allergy Immunol 2024 (PMID 38913142)
This is a shared conversation. Sign in to Orris to start your own chat.