Lymphophagocytic astrocytosis
Reading File
Checking Date & Time
Finding Sources
Finding Sources
Finding Sources
Searching PubMed
lymphophagocytic astrocytosis
Finding Sources
Searching the Web
lymphophagocytic astrocytosis definition pathology
Searching the Web
"lymphophagocytic astrocytosis" neuropathology
Finding Sources
Reading Article
PMID: 30726926
Finding Sources
Lymphophagocytic histiocytosis
Finding Sources
Finding Sources
Searching PubMed
"hemophagocytic lymphohistiocytosis"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Here is a comprehensive overview of Hemophagocytic Lymphohistiocytosis (HLH):
Hemophagocytic Lymphohistiocytosis (HLH)
Definition
HLH is a life-threatening syndrome of massive, uncontrolled hyperinflammation driven by unrelenting activation of CD8+ cytotoxic T lymphocytes and macrophages, leading to multiorgan damage — particularly in the liver, bone marrow, and CNS. It exists in a primary (genetic) and a secondary (acquired) form with a convergent clinical phenotype resembling septic shock.
— Robbins, Cotran & Kumar Pathologic Basis of Disease; Harrison's 22E
Classification
Primary HLH (Mendelian/Familial)
Caused by homozygous or compound heterozygous null mutations in genes controlling cytotoxic granule synthesis, trafficking, and function:
| Gene Defect | Syndrome |
|---|---|
| PRF1 (perforin) | FHL2 — ~30% of familial cases |
| UNC13D (Munc13-4) | FHL3 — ~30% |
| STX11 (syntaxin 11) | FHL4 — ~10% |
| STXBP2 (Munc18-2) | FHL5 — ~20% |
| SH2D1A | XLP1 |
| BIRC4 (XIAP) | XLP2 |
| RAB27A | Griscelli syndrome type 2 |
| LYST | Chédiak-Higashi syndrome |
Secondary HLH (Acquired)
- Infection-associated: Viruses (~70% of secondary cases) — EBV most common (40% of viral cases); also CMV, hepatitis viruses, influenza, COVID-19; bacteria (TB, Staphylococcus, Rickettsia); parasites (visceral leishmaniasis, malaria); fungi (Histoplasma)
- Malignancy-associated: Lymphoma is the trigger in >50% of adult cases
- Autoimmune-associated (MAS-HLH): Systemic JIA, adult-onset Still's, SLE, vasculitis
- Transplant-associated
Primary HLH mostly affects children; secondary HLH is far more common in adults.
— Harrison's 22E, Goldman-Cecil Medicine
Pathobiology
Under normal conditions, CTLs and NK cells release perforin-containing cytolytic granules to kill antigen-expressing target cells → target cell elimination → downregulation of the immune response (activation-induced cell death).
In HLH, this cytotoxic mechanism fails. Infected or antigen-bearing cells persist, causing continuous stimulation of CD8+ T cells and NK cells. These cells then switch to their secondary effector mechanism — massive cytokine secretion, especially IFN-γ, which is the master activator of macrophages. The result:
- Uncontrolled macrophage activation → hemophagocytosis (macrophages engulf RBCs, WBCs, platelets, and their precursors)
- Massive release of TNF-α, IL-6, IL-12, and IL-18 → cytokine storm
- Systemic inflammatory response → multiorgan failure
In adults with secondary HLH, ~15% harbor heterozygous hypomorphic alleles in familial HLH genes (most commonly the PRF1 A92V polymorphism, present in 5–8% of the general population), which may predispose to secondary HLH under a "second hit."
— Goldman-Cecil Medicine; Robbins Pathologic Basis of Disease
Clinical Features
| Feature | Details |
|---|---|
| Fever | High, unremitting; nearly universal |
| Hepatosplenomegaly | Prominent in both children and adults |
| Cytopenias | Bicytopenia required (thrombocytopenia most frequent; also anemia, neutropenia) |
| Coagulopathy | Low fibrinogen, DIC, severe bleeding risk |
| Liver involvement | Elevated transaminases, conjugated hyperbilirubinemia, elevated GGT; may mimic acute liver failure |
| CNS involvement | ~1/3 of children: seizures, decreased consciousness, meningismus, cranial nerve palsies; less common in adults |
| Rash | Non-specific maculopapular, petechiae, purpura |
| Lymphadenopathy | ~50% of children |
Children (primary HLH): Median onset 3–6 months; prominent CNS involvement; median survival without treatment 1–2 months.
Adults (secondary HLH): Present at any age; more often sepsis-like picture; CNS involvement less frequent; lymphoma is the most common trigger.
Diagnosis (HLH-2004 Criteria)
Diagnosis requires molecular diagnosis of familial disease OR ≥5 of these 8 criteria:
- Fever
- Splenomegaly
- Cytopenias of ≥2 lineages (Hgb <90 g/L, platelets <100×10⁹/L, neutrophils <1×10⁹/L)
- Hypertriglyceridemia (fasting TG ≥3.0 mmol/L) and/or hypofibrinogenemia (≤1.5 g/L)
- Hemophagocytosis on bone marrow, spleen, lymph node, or liver biopsy
- Low/absent NK cell activity
- Hyperferritinemia (ferritin ≥500 μg/L)
- Elevated soluble CD25 (sIL-2R) ≥2,400 U/mL
Key caveat: Hemophagocytosis in the bone marrow is neither sufficient nor required — it may be absent early in the course. Serial examinations may be needed. In sepsis vs. HLH, IFN-γ is markedly elevated in HLH while IL-6 dominates in sepsis — a useful distinguishing cytokine pattern.
— Goldman-Cecil Medicine; Harrison's 22E
Treatment
Algorithm:
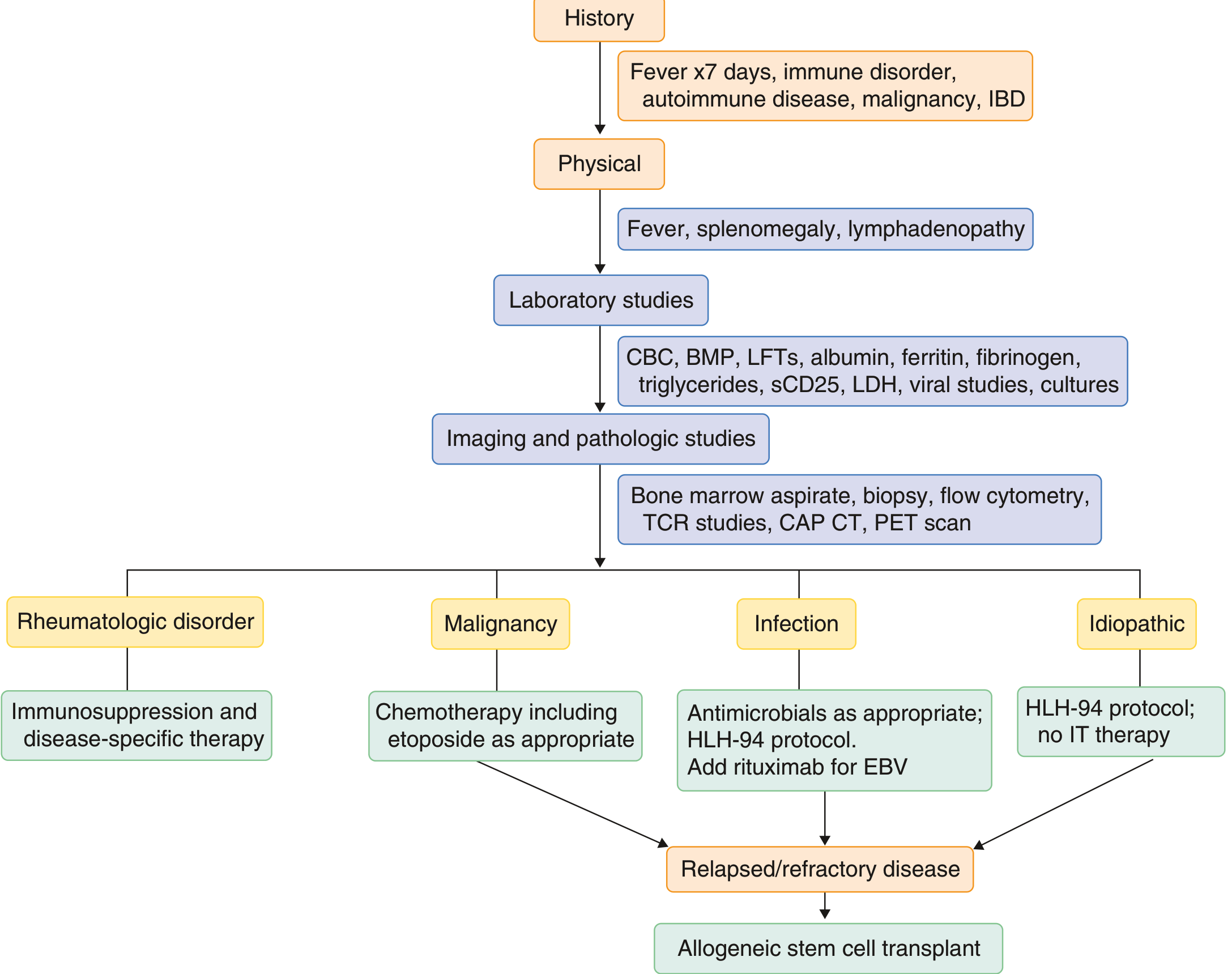
HLH-94 Protocol (first-line for primary/severe HLH):
- Dexamethasone 10 mg/m²/day × 2 weeks, tapering over 8 weeks
- Etoposide 150 mg/m² twice weekly × 2 weeks, then weekly
- Intrathecal methotrexate (for neurological involvement)
- Cyclosporine A added in HLH-2004 (from day 1, trough ~200 μg/L)
Secondary HLH — treat the underlying trigger:
- Malignancy: Etoposide-containing chemotherapy immediately
- EBV-HLH: Mild/moderate → glucocorticoids ± IVIG ± cyclosporine; severe → etoposide + dexamethasone + rituximab (anti-CD20); etoposide dosing less intense than in primary HLH (75–100 mg/m² weekly in age <40; 50–75 mg/m² weekly in age ≥40)
- Autoimmune/MAS: Immunosuppression + disease-specific therapy
- Emerging therapies: JAK1 inhibitors (targeting the IFN-γ pathway), IL-1/IL-6 inhibitors, emapalumab (anti-IFN-γ)
Curative therapy: Allogeneic HSCT — mandatory for familial HLH and refractory/relapsed disease. Without treatment, familial HLH is uniformly fatal. With treatment ± HSCT, ~50% survival, though neurologic sequelae (psychomotor retardation, epilepsy) affect ~1/3 of survivors.
— Harrison's 22E; Goldman-Cecil Medicine; Robbins Basic Pathology
Key Points to Remember
- HLH is a clinical syndrome, not a disease — always search for the underlying trigger
- The unifying mechanism is failure of CTL/NK cytotoxicity → IFN-γ-driven macrophage activation
- Ferritin >500 μg/L is sensitive but not specific; ferritin >10,000 μg/L is highly suggestive
- sCD25 (sIL-2R) is a powerful biomarker of disease activity
- In critically ill patients who don't respond to antibiotics, think HLH
- EBV is the single most common trigger across all forms
- JAK inhibition (targeting JAK1/IFN-γ axis) is an emerging treatment strategy
This is a shared conversation. Sign in to Orris to start your own chat.