Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND mechanism
Reading File
Reading File
Searching Images
ARDS pathogenesis alveolar capillary membrane neutrophil diagram
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have rich mechanistic content from the textbooks plus excellent diagrams. Let me compose a comprehensive answer.
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
ARDS is a syndrome of acute inflammatory lung injury characterized by non-cardiogenic pulmonary edema, severe hypoxemia (PaO₂/FiO₂ ≤ 300 mmHg), bilateral chest infiltrates, and onset within one week of a known precipitant. Its mechanism is best understood through three overlapping layers: the initial insult, cellular and molecular injury to the alveolar-capillary membrane, and the downstream consequences of barrier failure.
1. Precipitating Insults
ARDS arises from either direct (pulmonary) or indirect (extrapulmonary) injury:
| Direct | Indirect |
|---|---|
| Pneumonia, aspiration, inhalation injury, pulmonary contusion | Sepsis, severe pancreatitis, blood transfusions (TRALI), trauma, hemorrhage/hypotension |
Sepsis is the most common precipitant. Regardless of the trigger, the resulting lung injury is histologically identical — diffuse alveolar damage (DAD).
2. The Alveolar-Capillary Membrane — The Central Target
The alveolar-capillary unit consists of the alveolar epithelium (primarily Type I pneumocytes for gas exchange, Type II for surfactant production), the interstitium, and the microvascular endothelium. ARDS results from injury to both sides:
- Endothelial injury: Loss of pulmonary vascular endothelial barrier integrity is both necessary and sufficient for ARDS development. Endothelial cells become damaged via direct insult, cytokine-mediated apoptosis, and neutrophil-induced injury, causing tight junction disruption and massive increases in vascular permeability.
- Epithelial injury: Type I pneumocyte death (via necrosis, apoptosis, coagulation, and mechanical stretch) disrupts the epithelial barrier and impairs alveolar fluid clearance. Type II cell injury reduces surfactant production.
Murray & Nadel's Textbook of Respiratory Medicine, p. 3146
3. The Neutrophil — Central Cellular Mediator
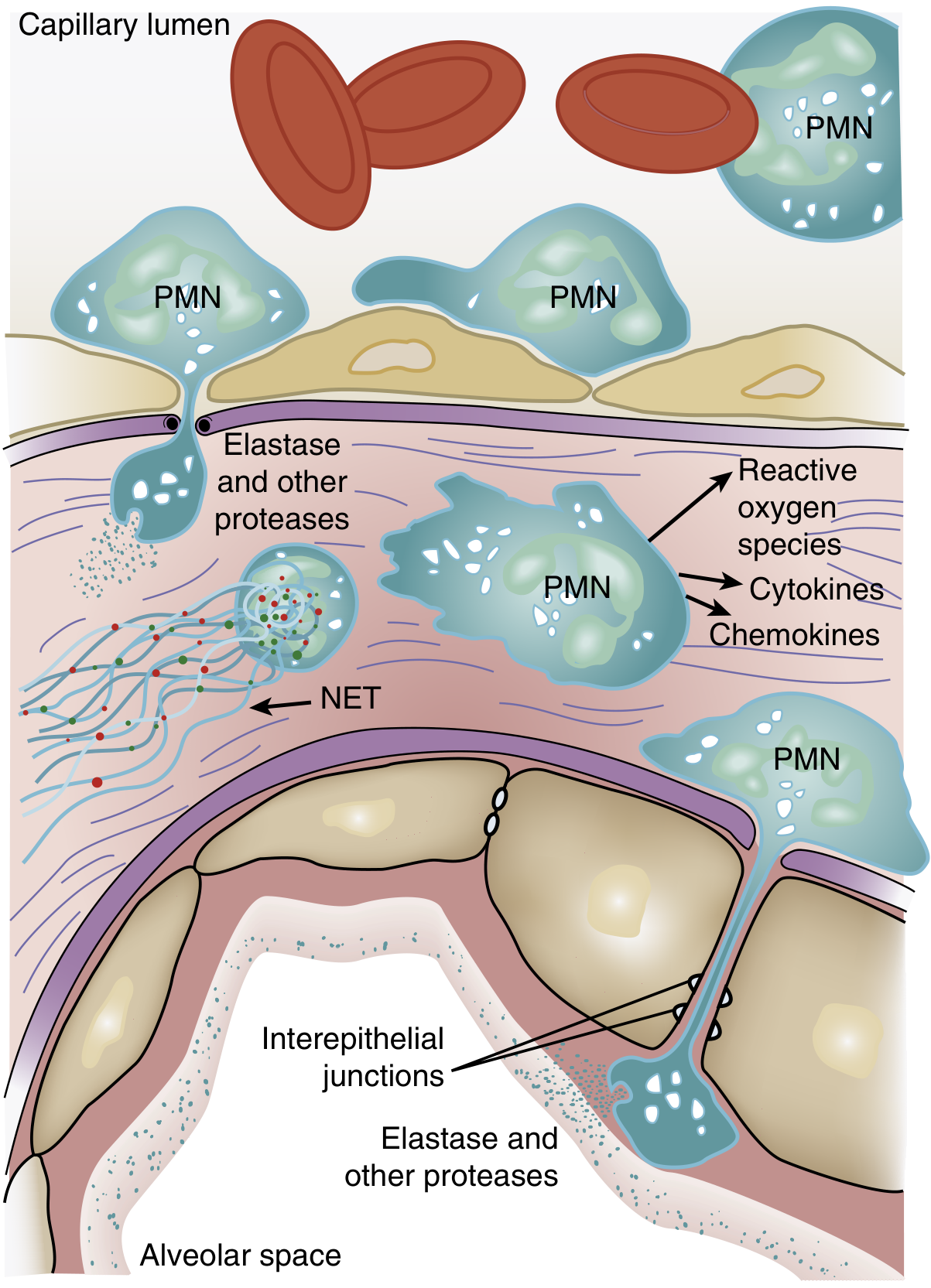
Neutrophil accumulation in the pulmonary microvasculature is one of the earliest and most characteristic features of ARDS:
Step-by-step neutrophil cascade:
- Sequestration: The average pulmonary capillary is narrower than a neutrophil. Activated neutrophils stiffen (due to actin cytoskeleton remodeling) and cannot deform adequately, becoming physically trapped in capillary segments. This produces a transient leukopenia — one of the earliest clinical signs.
- Transmigration: Sequestered neutrophils migrate across the endothelium into the interstitium and alveolar space, aided by chemokines (e.g., IL-8) released by macrophages and epithelial cells. Early migration can occur even without classic adhesion molecules (L-selectin, β2-integrins).
- Tissue destruction: Once in the alveolar space, activated PMNs release:
- Reactive oxygen species (ROS) — oxidative damage to membranes
- Proteolytic enzymes (elastase, matrix metalloproteinases) — degrade extracellular matrix and interepithelial junctions, and directly degrade surfactant protein A
- Cytokines: TNF-α, IL-1β — amplify the inflammatory response and recruit additional cells
- Neutrophil extracellular traps (NETs) — chromatin-based structures that trap pathogens but also promote tissue injury and coagulopathy
- Neutrophil–platelet interaction: Mutual activation of neutrophils and platelets amplifies both inflammation and microvascular thrombosis.
Murray & Nadel's, pp. 3147–3148
4. Cytokines, Macrophages, and the Inflammatory Cascade
Alveolar macrophages are the first responders to lung injury. They:
- Recognize pathogen-associated (PAMPs) and damage-associated (DAMPs) molecular patterns via toll-like receptors (TLRs)
- Release TNF-α, IL-1β, IL-6, IL-8, and platelet-activating factor (PAF)
- Activate the p38 MAP kinase pathway, further amplifying TNF-α production and macrophage inflammatory protein-2 (a neutrophil chemokine)
This cytokine storm creates a self-perpetuating feed-forward loop: macrophages activate neutrophils, neutrophils damage tissue and release more cytokines, more neutrophils are recruited.
5. Surfactant Dysfunction
Type II pneumocyte injury and the inflammatory milieu cause:
- Decreased production of surfactant (especially dipalmitoylphosphatidylcholine)
- Shift from large (active) to small (inactive) surfactant aggregates
- Plasma protein leak into alveoli, which inhibits surfactant function
- Elastase-mediated degradation of surfactant protein A
The result: alveolar units collapse (atelectasis), worsening ventilation-perfusion (V/Q) mismatch and hypoxemia. Unlike neonatal RDS, adult surfactant supplementation trials have consistently failed to reduce mortality.
6. Angiopoietin Axis and Vascular Permeability
- Angiopoietin-1 (Ang1) stabilizes endothelial tight junctions via the Tie2 receptor.
- Angiopoietin-2 (Ang2) is released by injured endothelial cells as a competitive antagonist, disrupting barrier integrity. Elevated plasma Ang2 is a biomarker of severity in sepsis and ARDS. Genetic variants in Ang2 are associated with increased ARDS risk.
Murray & Nadel's, p. 3149
7. Failure of Alveolar Fluid Clearance
Normally, Na⁺ channels (ENaC) on apical epithelial surfaces drive sodium into the cell, with water following osmotically. Na⁺/K⁺-ATPase on the basolateral surface maintains the gradient. In ARDS:
- Hypoxia suppresses ENaC expression and Na⁺/K⁺-ATPase activity
- Nitric oxide (from inflammatory activation) impairs β-adrenergic-mediated fluid clearance upregulation
This means even if permeability is restored, the lung cannot actively clear the protein-rich edema fluid. Attempts to pharmacologically boost fluid clearance with β2-agonists have been disappointing in clinical trials.
8. Phases of ARDS
| Phase | Timing | Pathology |
|---|---|---|
| Exudative | Days 1–7 | Diffuse alveolar damage, hyaline membrane formation, protein-rich edema, neutrophil infiltration |
| Proliferative | Days 7–21 | Type II pneumocyte hyperplasia, fibroblast infiltration, early fibrosis, organizing exudate |
| Fibrotic | Weeks–months | Progressive fibrosis, architectural distortion; associated with prolonged ventilation dependence |
9. Ventilator-Induced Lung Injury (VILI) — A Perpetuating Mechanism
Mechanical ventilation, while life-sustaining, can worsen ARDS through:
- Volutrauma/Barotrauma: Overdistension of aerated alveoli at high tidal volumes → epithelial and endothelial tears
- Atelectrauma: Repetitive opening and collapse of unstable alveoli at low end-expiratory volumes
- Biotrauma: Mechanical stretch triggers cytokine release (IL-6, IL-8, TNF-α) from the lung into the systemic circulation → distal organ dysfunction
This is the mechanistic basis for lung-protective ventilation (6 mL/kg IBW, plateau pressure ≤30 cmH₂O, adequate PEEP).
Summary Diagram of Mechanism
Precipitant (sepsis, aspiration, trauma)
↓
Macrophage activation → Cytokine storm (TNF-α, IL-1β, IL-8)
↓
Neutrophil sequestration in pulmonary capillaries
↓
PMN transmigration + activation
↓
ROS + Elastase + NETs → Alveolar-capillary barrier destruction
↓
Protein-rich edema floods alveoli
↓
Surfactant dysfunction + Type I/II pneumocyte death
↓
Alveolar collapse (atelectasis) + impaired fluid clearance
↓
Severe V/Q mismatch → Refractory hypoxemia (ARDS)
↓
Mechanical ventilation → VILI → Perpetuates injury
The net result is a diffusely stiff, edematous, non-homogeneous lung with severely impaired gas exchange, requiring mechanical ventilatory support until the alveolar-capillary barrier recovers — or progressing to fibrosis.
Sources: Murray & Nadel's Textbook of Respiratory Medicine, pp. 3081–3230; Goldman-Cecil Medicine, p. 1065; Sabiston Textbook of Surgery, p. 940
This is a shared conversation. Sign in to Orris to start your own chat.