Explain biochemistry of gastric disorders in detail for a first year MBBS student
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have comprehensive, authoritative content from multiple major textbooks. Let me compile the full detailed answer.
Biochemistry of Gastric Disorders - A Guide for First Year MBBS Students
1. OVERVIEW: The Stomach as a Biochemical Organ
The stomach serves multiple biochemical functions: temporary storage of ingested food, mechanical and chemical digestion, controlled delivery of chyme to the duodenum, protection against ingested pathogens, and facilitation of iron and calcium absorption. Understanding its normal biochemistry is the foundation for understanding what goes wrong in gastric disorders.
The stomach has four anatomical-functional regions with distinct cell populations:
- Fundus/Corpus (body): Contains oxyntic glands with parietal cells (HCl + intrinsic factor), chief cells (pepsinogen), and ECL cells (histamine).
- Antrum/Pylorus: Contains pyloric glands with G cells (gastrin) and D cells (somatostatin).
2. GASTRIC ACID SECRETION - THE CORE BIOCHEMISTRY
2.1 The Parietal Cell and the Proton Pump
The parietal cell is the master acid-secreting cell. Each parietal cell secretes approximately 3 × 10⁴ hydrogen ions per second, producing HCl at a final concentration of ~150 mmol/L. This is about 1 million times the hydrogen ion concentration of blood (pH 7.4 vs. pH ~1 in the gastric lumen) - one of the steepest electrochemical gradients in the human body.
The enzyme responsible is H⁺/K⁺-ATPase (the proton pump):
H⁺ is pumped OUT into the gastric lumen in exchange for K⁺ being pumped IN, using ATP hydrolysis.
- Inside the parietal cell: CO₂ + H₂O → H₂CO₃ (catalyzed by carbonic anhydrase)
- H₂CO₃ dissociates → H⁺ + HCO₃⁻
- H⁺ is exported into the lumen via H⁺/K⁺-ATPase
- HCO₃⁻ leaves the cell into blood via Cl⁻/HCO₃⁻ exchanger on the basolateral side - this is the "alkaline tide" (blood becomes slightly alkaline after a meal)
- Cl⁻ is secreted via CFTR and SLC26A7 channels into the lumen, combining with H⁺ to form HCl
When the parietal cell is stimulated, cytoplasmic vesicles containing H⁺/K⁺-ATPase fuse with the apical membrane, dramatically expanding the secretory canalicular surface area and positioning the proton pumps to maximise acid secretion. - Yamada's Textbook of Gastroenterology, 7th ed.
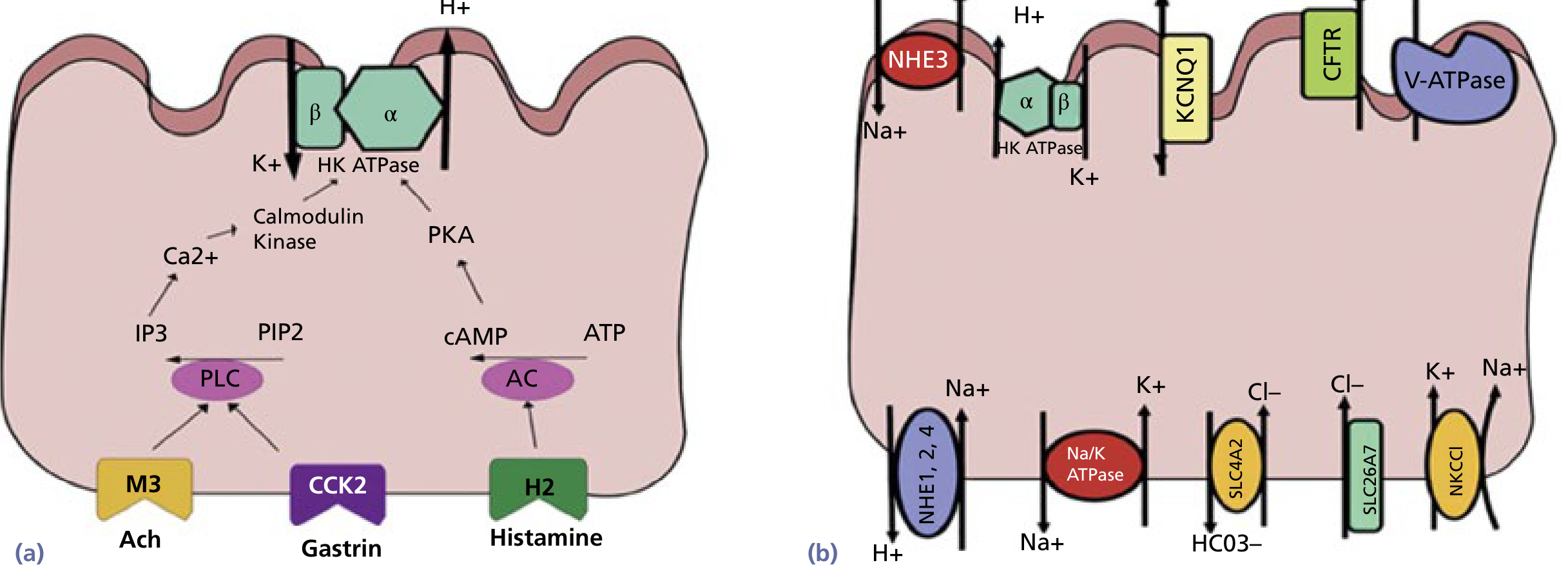
2.2 Three Stimulatory Pathways (The "Holy Trinity" of Acid Stimulation)
Three major stimulants act on three distinct receptors on the parietal cell:
| Stimulant | Source | Receptor | Signal Pathway |
|---|---|---|---|
| Acetylcholine (ACh) | Vagus nerve / enteric neurons | M3 (muscarinic) | PLC → IP3 → ↑Ca²⁺ → Calmodulin kinase → H⁺/K⁺-ATPase |
| Gastrin | G cells of gastric antrum | CCK-2 (CCK subtype B) | PLC → IP3 → ↑Ca²⁺ → H⁺/K⁺-ATPase |
| Histamine | ECL cells (enterochromaffin-like) | H2 | Adenylyl cyclase → ↑cAMP → PKA → H⁺/K⁺-ATPase |
Key concept: Histamine is the most important paracrine stimulant. Gastrin and ACh primarily stimulate ECL cells to release histamine, which then acts on parietal cells. This is why H2-blockers (like cimetidine, ranitidine) and proton pump inhibitors (PPIs) are so effective - they block the most powerful proximal or final common pathway. - Bailey and Love's Short Practice of Surgery, 28th Ed.
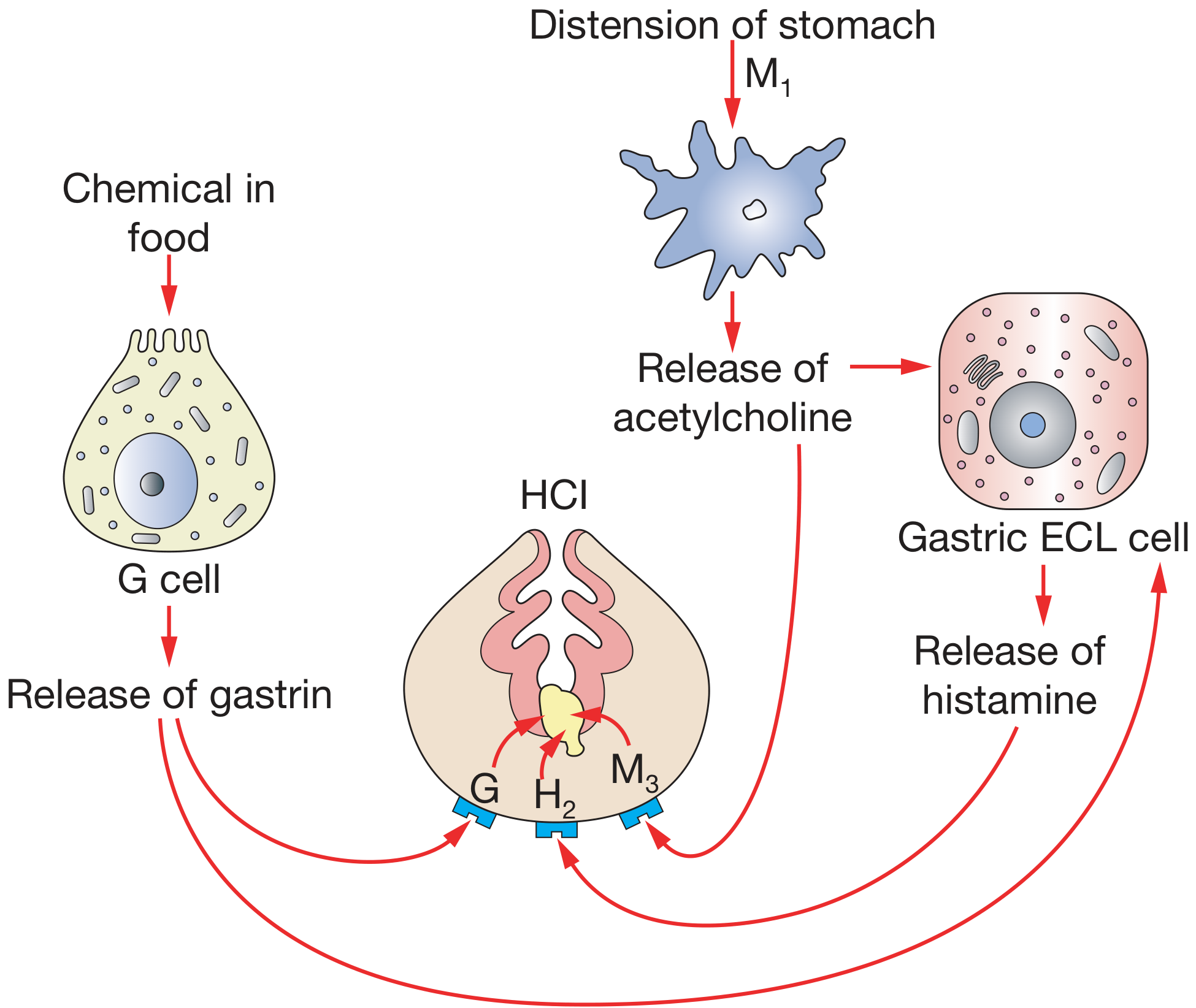
3. THREE PHASES OF GASTRIC SECRETION
| Phase | Trigger | Mechanism | % of Total Acid |
|---|---|---|---|
| Cephalic | Sight, smell, taste, thought of food | Vagus nerve → ACh → parietal cell + G cells | ~30% |
| Gastric | Food in stomach (distension + chemicals) | Gastrin from G cells; local neural reflexes | ~60% |
| Intestinal | Chyme entering duodenum | Initially small stimulation; then inhibition by secretin, CCK, GIP | ~10% (net inhibitory) |
Negative feedback / inhibitory loop:
- When luminal pH falls below ~3.0, acid itself inhibits G cell secretion of gastrin (direct inhibition)
- Acidification also stimulates D cells to release somatostatin, which inhibits G cells, ECL cells, and parietal cells
- Duodenal acidification releases secretin from S cells → inhibits gastric acid and stimulates pancreatic bicarbonate
- Other intestinal inhibitors: GIP (gastric inhibitory polypeptide), VIP
4. PEPSIN AND PROTEOLYTIC DIGESTION
Chief cells (also called zymogenic cells) in the oxyntic glands secrete pepsinogen - an inactive zymogen (precursor). This is a key biochemical safety mechanism: storing a proteolytic enzyme in inactive form prevents autodigestion.
Activation of pepsinogen:
- Pepsinogen (inactive, MW ~42 kDa) is secreted into the acidic gastric lumen
- At pH < 5, HCl cleaves an inhibitory peptide from the N-terminus → active pepsin (MW ~35 kDa)
- Pepsin is autocatalytic - it can activate more pepsinogen at pH 1-3 (optimal pH for pepsin activity)
- At pH > 7, pepsin is irreversibly denatured (this protects the duodenum/small intestine)
Function of pepsin:
- Cleaves peptide bonds adjacent to aromatic amino acids (Phe, Tyr, Trp) - an endopeptidase
- Initiates protein digestion, breaking large proteins into smaller polypeptides
- Digests ~15-20% of dietary protein; the majority is completed in the small intestine by pancreatic proteases
5. THE GASTRIC MUCOSAL BARRIER - DEFENCE MECHANISMS
The stomach must protect itself from its own corrosive acid (pH 1-2) and pepsin. Several defence layers work in concert:
5.1 Mucus Layer
- Produced by surface mucous cells (lining cells) and pyloric gland mucous cells
- A viscid gel of mucopolysaccharides (glycoproteins), primarily mucin
- Forms a 0.2-0.5 mm unstirred layer on the mucosal surface
- Acts as a physical barrier and has considerable buffering capacity (enhanced by bicarbonate ions within the mucus)
5.2 Bicarbonate Secretion
- Surface epithelial cells actively secrete HCO₃⁻ into the mucus layer
- Creates a pH gradient: pH ~1-2 at the luminal surface, pH ~7 at the epithelial cell surface
- Prostaglandins (particularly PGE₂ and PGI₂) stimulate both mucus and bicarbonate secretion
5.3 Epithelial Cell Tight Junctions
- Prevent back-diffusion of H⁺ from the lumen into the mucosa
- Rapid cell turnover (every 3-5 days) repairs any breaches
5.4 Mucosal Blood Flow
- Rich submucosal blood supply washes away any acid that penetrates the barrier and delivers oxygen and nutrients for rapid repair
Factors that break down the mucosal barrier:
- NSAIDs (inhibit COX enzymes → reduced prostaglandin synthesis → reduced mucus and bicarbonate)
- Bile reflux
- Alcohol
- H. pylori infection
- Ischaemia/shock (the stomach is the most sensitive part of the GIT to ischaemia and the slowest to recover - explaining stress ulcers in critically ill patients) - Bailey and Love's Short Practice of Surgery
6. HELICOBACTER PYLORI - BIOCHEMICAL PATHOGENESIS
H. pylori is the most important biochemical disruptor of the stomach. It is a gram-negative, spiral-shaped bacterium with polar flagella.
6.1 How H. pylori Survives the Acid Environment
The organism would be killed by the acidic lumen. Its survival strategies:
- Urease enzyme: H. pylori produces large amounts of urease
- Urea (diffusing from blood) → NH₃ + CO₂ (catalyzed by urease)
- NH₃ neutralizes surrounding acid, creating a microenvironment at pH ~6-7 around the organism
- This is the basis of the urea breath test (patient drinks ¹³C-labeled urea; if H. pylori is present, ¹³C-CO₂ is detected in expired breath)
- Motility: Flagella allow it to move away from the acidic lumen toward the mucus layer, following a pH gradient
- Adhesins: >30 outer membrane proteins allow it to adhere to gastric epithelial cells
6.2 Virulence Factors
VacA (Vacuolating Cytotoxin A):
- A pore-forming cytotoxin
- Forms a ring structure (flower shape) that inserts into the host cell membrane, creating ion channels
- Channels allow leakage of urea and nutrients out of cells (feeding the bacterium)
- Also inserts into endosomal membranes → cellular vacuolation and swelling
- Damages mitochondrial membranes → apoptosis
CagA (Cytotoxin-Associated Gene A):
- Part of a pathogenicity island - a stretch of DNA inserted into virulent strains' genome
- Adjacent genes encode a Type IV secretion system - a molecular needle that injects CagA directly into host gastric cells
- Inside host cells, CagA is phosphorylated on tyrosine residues by host kinases (SFKs)
- Phospho-CagA activates multiple signalling pathways:
- Disrupts cell polarity and cytoskeletal function
- Disrupts apical junctions between epithelial cells → gaps in the epithelium → increased mucosal permeability
- Activates growth factor receptor-like signalling → abnormal cell proliferation (a mechanism for gastric carcinogenesis)
- Elicits a vigorous inflammatory response
6.3 How H. pylori Causes Peptic Ulcers
- ~75% of all peptic ulcer disease (PUD) is caused by H. pylori
- H. pylori infection → antral gastritis → increased gastrin secretion (G cells are stimulated, while somatostatin from D cells is suppressed) → increased acid output
- Bacterial toxins (VacA, CagA) directly damage mucosal cells → breakdown of the mucosal barrier
- The combination of excess acid + breached barrier = ulceration
- Only ~15% of infected individuals develop peptic ulcers - individual virulence factor expression (CagA+, VacA+) and host genetic susceptibility determine outcome - Mulholland and Greenfield's Surgery, 7th Ed.
7. NSAIDs AND GASTRIC ULCER BIOCHEMISTRY
This is the second major cause of PUD after H. pylori.
Mechanism (two-pronged attack):
-
Direct local injury: NSAIDs are weak acids (e.g. aspirin, ibuprofen). In the acidic stomach (pH 1-2), they remain in non-ionized lipid-soluble form → penetrate the mucosal cell membranes → ionize inside the neutral pH cytoplasm → become trapped (ion trapping) → cause direct cellular damage
-
Systemic COX inhibition (the more important mechanism):
- NSAIDs inhibit cyclooxygenase (COX-1) enzyme
- COX-1 is constitutively expressed in gastric mucosa and converts arachidonic acid → prostaglandins (PGE₂, PGI₂)
- Prostaglandins normally: stimulate mucus secretion, stimulate bicarbonate secretion, maintain mucosal blood flow, promote epithelial cell proliferation
- When COX-1 is inhibited → ↓ prostaglandins → ↓ mucus → ↓ bicarbonate → reduced blood flow → impaired repair → ulceration
- COX-2 selective NSAIDs (e.g. celecoxib) spare COX-1 → less gastric toxicity
8. SPECIFIC GASTRIC DISORDERS AND THEIR BIOCHEMISTRY
8.1 Zollinger-Ellison Syndrome (Gastrinoma)
- A gastrin-secreting tumour (gastrinoma), usually in the pancreas or duodenum
- Causes massively elevated serum gastrin → continuous, maximal stimulation of parietal cells
- Results in: severe peptic ulcers (multiple, in unusual locations), watery diarrhoea (acid inactivates pancreatic lipase → steatorrhoea), and oesophageal ulceration
- The excess HCl cannot be neutralized in the duodenum
- Biochemical diagnosis: fasting serum gastrin > 1000 pg/mL + basal acid output > 15 mEq/hr; secretin stimulation test - normally secretin inhibits gastrin, but in gastrinoma it paradoxically increases it
8.2 Pernicious Anaemia (Autoimmune Gastritis)
- Autoantibodies against H⁺/K⁺-ATPase (the proton pump, a 92-kDa protein on the parietal cell luminal membrane) → parietal cell destruction
- Intrinsic factor (IF) is also produced exclusively by parietal cells; its destruction → IF deficiency → vitamin B12 malabsorption → megaloblastic anaemia
- Anti-intrinsic factor antibodies (Type I = blocking antibodies, Type II = binding antibodies) are more specific for pernicious anaemia
- Chronic achlorhydria → G cells release gastrin excessively (loss of negative feedback) → hypergastrinaemia → ECL cell hyperplasia (risk of gastric carcinoid tumour)
8.3 Stress Ulcers (Acute Stress Gastritis)
- Occur in critically ill patients (burns - Curling's ulcer; brain injury - Cushing's ulcer; sepsis, trauma)
- The stomach is the most sensitive organ in the GIT to ischaemia, and the slowest to recover (demonstrated by tonometry studies)
- Ischaemia → reduced mucosal blood flow → failure to maintain the mucosal barrier → back-diffusion of H⁺ → mucosal necrosis
- Curling's ulcers (burns): hypovolaemia + ischaemia; Cushing's ulcers (brain injury): vagal overstimulation → excess acid secretion
- Prevention: early enteral feeding, PPIs/H2 blockers for stress ulcer prophylaxis
8.4 Achlorhydria and Hypochlorhydria
- Absence or reduction of gastric acid
- Causes: H. pylori-associated atrophic gastritis, autoimmune gastritis (pernicious anaemia), PPI use
- Consequences:
- Impaired pepsinogen activation → reduced protein digestion
- Reduced iron absorption (ferric → ferrous requires acid)
- Reduced calcium absorption
- Bacterial overgrowth (acid normally kills ingested organisms)
- Vitamin B12 malabsorption (requires acid to release B12 from dietary proteins)
9. DRUGS ACTING ON GASTRIC BIOCHEMISTRY
| Drug Class | Example | Mechanism of Action |
|---|---|---|
| Proton Pump Inhibitors (PPIs) | Omeprazole, pantoprazole | Irreversibly bind H⁺/K⁺-ATPase in the secretory canaliculus (prodrug activated at pH < 5) → block final common pathway of acid secretion |
| H2 Receptor Antagonists | Ranitidine, famotidine | Competitively block H2 receptors on parietal cells → reduce histamine-stimulated cAMP → reduce acid |
| Antacids | Aluminium hydroxide, Mg(OH)₂ | Neutralize secreted HCl directly (not biochemical inhibition) |
| Misoprostol | Misoprostol | Synthetic PGE₁ analogue → stimulates mucus/bicarbonate secretion + reduces acid; used with NSAIDs |
| H. pylori eradication | Triple/quad therapy | Antibiotics (clarithromycin, amoxicillin, metronidazole) + PPI |
PPIs - mechanism in detail:
PPIs (substituted benzimidazoles) are weak bases (pKa 4-5). At blood pH 7.4, they are non-ionized and membrane-permeable - they reach the parietal cell. In the highly acidic secretory canaliculus (pH ~1), they become protonated and trapped, and convert to the active sulphenamide form - which then covalently binds (irreversibly) to cysteine residues on H⁺/K⁺-ATPase. Acid secretion only resumes when new pump proteins are synthesized (~18-24 hours) - Yamada's Textbook of Gastroenterology, 7th Ed.
10. QUICK SUMMARY TABLE - KEY CELLS AND SECRETIONS
| Cell Type | Location | Product | Stimulus | Function |
|---|---|---|---|---|
| Parietal cell | Oxyntic glands (body/fundus) | HCl + Intrinsic Factor | ACh, Gastrin, Histamine | Protein digestion, B12 absorption, kills microbes |
| Chief cell | Oxyntic glands | Pepsinogen | ACh, secretin | Activated to pepsin by HCl; protein digestion |
| G cell | Antrum | Gastrin | Food proteins, distension, vagus | Stimulates acid + ECL cells |
| ECL cell | Oxyntic glands | Histamine | Gastrin, ACh | Paracrine stimulation of parietal cells |
| D cell | Antrum + Corpus | Somatostatin | Acid, vagal stimulation | Inhibits G cells, ECL cells, parietal cells (negative feedback) |
| Mucous cells | All gastric mucosa | Mucus (glycoproteins) | Prostaglandins, ACh | Mucosal barrier |
| S cell (duodenum) | Duodenum | Secretin | Duodenal acid | Inhibits gastric acid; stimulates pancreatic bicarbonate |
KEY EXAM POINTS FOR FIRST YEAR MBBS
- The proton pump (H⁺/K⁺-ATPase) is the final common pathway of acid secretion - PPIs block it.
- Pepsinogen requires acidic pH < 5 for activation to pepsin; pepsin works optimally at pH 1-3.
- The cephalic phase is vagally mediated (conditioned reflex, as shown by Pavlov's experiments).
- H. pylori survives by producing urease (urea → NH₃ neutralizes acid locally).
- VacA is a pore-forming toxin; CagA is a phosphorylated virulence factor injected via a Type IV secretion system.
- NSAIDs cause ulcers primarily by inhibiting COX-1 → reducing gastric prostaglandins.
- Pernicious anaemia = anti-parietal cell antibodies → loss of HCl + loss of intrinsic factor → B12 deficiency.
- Zollinger-Ellison syndrome = gastrinoma → hypergastrinaemia → excess acid → multiple ulcers.
- The "alkaline tide": after eating, blood becomes transiently alkaline because HCO₃⁻ leaves parietal cells into the blood in exchange for Cl⁻.
- Somatostatin from D cells is the major "off switch" for gastric acid - it inhibits G cells, ECL cells, and parietal cells simultaneously.
Sources: Bailey and Love's Short Practice of Surgery (28th Ed.), Yamada's Textbook of Gastroenterology (7th Ed.), Mulholland and Greenfield's Surgery (7th Ed.), Goodman & Gilman's Pharmacological Basis of Therapeutics, Henry's Clinical Diagnosis and Management by Laboratory Methods
This is a shared conversation. Sign in to Orris to start your own chat.