Some Important topics: Leukaemia, anemia, purpura, hodgkin lymphoma, thalassemia, sickle cell anemia, DIC, blood grouping. Everyone please study these topics now tomorrow i have exams on these topics teach me from Robbins
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is your complete Robbins-based exam study guide covering all 8 topics. Good luck tomorrow!
🩸 Haematology Exam Study Guide — From Robbins & Kumar Basic Pathology
1. ANEMIA
Definition: Reduction in RBC mass / haemoglobin below normal, impairing oxygen delivery.
Classification (Robbins):
| Type | Causes |
|---|---|
| Microcytic | Iron deficiency, thalassemia |
| Macrocytic | Folate / Vitamin B12 deficiency; elevated reticulocyte count |
| Normocytic | Hereditary spherocytosis, sickle cell disease |
Causes:
- Blood loss (hemorrhage)
- Increased destruction (hemolysis)
- Decreased production (marrow failure)
Hemolytic Anemias — Key Distinction:
Extravascular hemolysis (RBCs destroyed in spleen by macrophages):
- Hyperbilirubinemia + jaundice
- Splenomegaly
- Pigment gallstones (if long-standing)
Intravascular hemolysis (RBCs burst in circulation):
- Hemoglobinemia, hemoglobinuria, hemosiderinuria
- Iron deficiency (iron lost in urine)
- Decreased haptoglobin (seen in both types)
Clinical Features:
- Acute (large bleed): dyspnea, organ failure, shock
- Chronic: pallor, fatigue, lassitude
- Severe congenital: growth retardation, bone deformities from marrow hyperplasia (especially thalassemia)
Robbins & Kumar Basic Pathology, p.433
2. SICKLE CELL ANEMIA
Genetics: Autosomal recessive. Single amino acid substitution in β-globin - valine replaces glutamate at position 6. Creates Hb-S (HbS).
Epidemiology: Most common familial hemolytic anemia. HbS is protective against falciparum malaria - prevalent in equatorial Africa, parts of India, southern Europe, Middle East. ~8% of African Americans are HbS carriers; 1 in 600 have sickle cell anemia.
Pathogenesis:
- Deoxygenated HbS self-associates into polymers → distorts RBC into elongated crescentic/sickle shape
- Initially reversible on reoxygenation; repeated episodes cause membrane damage (Ca²⁺ influx, loss of K⁺/water) → irreversibly sickled cells prone to hemolysis
Factors determining clinical severity:
- Level of other haemoglobins (HbA, HbF): HbA retards sickling → heterozygotes (HbAS, "sickle cell trait") rarely sickle in vivo. HbF also inhibits sickling - newborns asymptomatic until HbF falls at ~5-6 months
- Degree of deoxygenation: low O₂ triggers sickling
- Intracellular Hb concentration: dehydration worsens sickling
Consequences:
- Hemolytic anemia (moderate to severe) - extravascular + intravascular
- Vaso-occlusion: sickled cells block microvessels → pain crises, tissue infarction (bones, spleen, lungs, brain)
- Splenic sequestration → functional asplenia by adulthood (autosplenectomy)
- Susceptibility to encapsulated bacteria (Streptococcus pneumoniae, H. influenzae) due to asplenia
- Stroke risk
- Aplastic crisis triggered by parvovirus B19
Robbins & Kumar Basic Pathology, p.384–386
3. THALASSEMIA
Definition: Inherited disorders causing decreased synthesis of α- or β-globin chains → Hb deficiency + intracellular precipitates of excess unpaired chains → hemolysis.
Genetics: Autosomal codominant. Common in Mediterranean, Africa, Asia (malaria belt).
- β-globin gene: chromosome 11 (single gene - mainly point mutations)
- α-globin genes: chromosome 16 (two tandem genes - mainly deletions)
Classification Table (Robbins):
| Syndrome | Genotype | Features |
|---|---|---|
| β-Thal Major | Homozygous (β⁰/β⁰ or β⁺/β⁺) | Severe anemia, requires regular transfusions |
| β-Thal Intermedia | Variable | Moderately severe, transfusions not always needed |
| β-Thal Minor | Heterozygous (β⁺/β or β⁰/β) | Asymptomatic/mild, red cell abnormalities on film |
| Silent carrier (α) | -/α, α/α | Asymptomatic, no red cell abnormality |
| α-Thal Trait | --/αα (Asian) or -/α, -/α (African) | Resembles β-thal minor |
| HbH Disease | --/-α | Moderately severe, β₄ tetramers (HbH) |
| Hydrops Fetalis | --/-- | Incompatible with life; Hb Barts (γ₄) |
Key Pathology:
- Excess unpaired α-chains precipitate → damage RBC membranes → extravascular hemolysis
- Ineffective erythropoiesis → marked marrow erythroid hyperplasia → bone deformities ("chipmunk face," "hair-on-end" skull X-ray in β-thal major)
- Iron overload (from transfusions + increased GI absorption) → heart and endocrine failure
Robbins & Kumar Basic Pathology, p.386–388
4. LEUKAEMIA
Leukaemia = malignant neoplasm of haematopoietic precursors that spill into the blood.
A. Acute Lymphoblastic Leukaemia/Lymphoma (ALL)
- Most common cancer in children. ~85% are B-ALL; T-ALL more common in adolescent males as thymic masses
- B-ALL peaks at age ~3 years; T-ALL peaks in adolescence
- Composed of immature B or T lymphoblasts
Pathogenesis:
- Mutations in transcription factors (B-ALL: PAX5, EBF1, TCF3, RUNX1, BCR-ABL1; T-ALL: NOTCH1)
- ~90% have chromosomal changes: most common is hyperploidy (>50 chromosomes) → good prognosis
- Philadelphia chromosome (BCR-ABL1) in ~5% of childhood ALL - poorer prognosis; treated with tyrosine kinase inhibitors
Clinical: lymphadenopathy, hepatosplenomegaly, bone pain, CNS involvement (headache, vomiting)
B. Chronic Myeloid Leukaemia (CML)
- Adults aged 25-60; peak incidence 4th-5th decade
- t(9;22) Philadelphia chromosome in ~95% → BCR-ABL fusion gene → constitutively active tyrosine kinase → growth factor-independent proliferation
- Affects granulocytic, erythroid, megakaryocytic, B-cell precursors (transformed haematopoietic stem cell)
Morphology:
- WBC often >100,000/μL with neutrophils, immature granulocytes, basophils, eosinophils
- Bone marrow hypercellular; massive splenomegaly (extramedullary haematopoiesis + splenic infarcts)
Clinical course:
- Insidious onset: fatigue, weakness, weight loss, dragging sensation in abdomen
- ~50% undergo blast crisis (transformation to AML or ALL) - median survival untreated = 3 years
- Treatment: Imatinib (BCR-ABL tyrosine kinase inhibitor) has transformed prognosis
C. Chronic Lymphocytic Leukaemia / Small Lymphocytic Lymphoma (CLL/SLL)
- Most common leukemia of adults in the Western world
- CLL: peripheral blood lymphocytes >5,000/μL; SLL: predominantly lymph node involvement (4% of NHLs)
- Pathogenesis: BCL2 overexpression (inhibits apoptosis; via deletion 13q) + BCR signaling via Bruton tyrosine kinase (BTK)
- Causes immune dysregulation → hypogammaglobulinemia + 15% develop warm autoantibodies (against own RBCs or platelets)
Morphology: Small resting lymphocytes with dark round nuclei; proliferation centers are pathognomonic
Treatment: BTK inhibitors (ibrutinib)
Robbins & Kumar Basic Pathology, p.411–412
Acute Myeloid Leukaemia (AML) — Brief:
- Aggressive tumor of immature myeloid blasts replacing marrow
- Mutations in transcription factors (interfere with myeloid differentiation) + growth factor receptor signaling or epigenome regulators
5. HODGKIN LYMPHOMA
Distinguishing features (Robbins):
- Distinctive Reed-Sternberg (RS) giant cells (neoplastic cells)
- Robust but ineffective host immune response - RS cells are minority of tumor mass
- Arises in single lymph node or chain - spreads in stepwise, contiguous fashion (unlike NHL)
Classification - 5 Subtypes:
| Subtype | Notes |
|---|---|
| Nodular Sclerosis | Most common (~70%); bands of collagen, lacunar RS cells; young adults, mediastinum |
| Mixed Cellularity | ~25%; EBV+ in up to 70%; classic RS cells; middle-aged |
| Lymphocyte Rich | Rare; good prognosis |
| Lymphocyte Depletion | Rare; worst prognosis; elderly/HIV |
| Nodular Lymphocyte Predominant | Different - expresses germinal center B-cell markers on RS cells |
The first four = Classic Hodgkin Lymphoma (share morphology and immunophenotype).
Pathogenesis:
- RS cells arise from germinal center B cells (proven by microdissection showing same immunoglobulin gene rearrangements with somatic hypermutation in every RS cell)
- EBV present in RS cells in up to 70% of mixed cellularity; identical EBV integration in all RS cells → infection precedes transformation
- RS cells secrete cytokines: IL-5 (eosinophil chemoattractant), TGF-β (fibrogenic), IL-13 (autocrine RS growth)
- RS cells escape immunity: loss of β₂-microglobulin (no MHC class I expression) + overexpression of PD-L1 (inhibits T-cell responses) - chromosome 9p amplification
Reed-Sternberg Cell Morphology:
- Giant binucleated/multilobed cell
- "Owl-eye" prominent eosinophilic nucleoli
- Immunophenotype: CD15+, CD30+, PAX5 dim, negative for most B-cell markers (CD20-)
Robbins & Kumar Basic Pathology, p.417–420
6. PURPURA / THROMBOCYTOPENIA
Thrombocytopenia = platelet count <150,000/μL
- Risk of post-traumatic bleeding: 20,000-50,000/μL
- Risk of spontaneous bleeding: <5,000/μL
- Bleeding is superficial: petechiae, ecchymoses, epistaxis, gum bleeding, mucosal hemorrhage
Causes (Robbins Table):
Decreased Production:
- Aplastic anemia, marrow infiltration (leukemia), drug-induced (alcohol, thiazides, cytotoxic drugs), infections (measles, HIV), megaloblastic anemia
Decreased Platelet Survival:
- Immunologic: ITP, SLE, drug-associated (quinidine, heparin), infections
- Non-immunologic: DIC, TTP, HUS, microangiopathic hemolytic anemias
Sequestration: Hypersplenism
Dilutional: Multiple transfusions
Immune Thrombocytopenic Purpura (ITP)
Two subtypes:
- Chronic ITP: Women aged 20-40; autoimmune
- Acute ITP: Children after viral infections; self-limited
Pathogenesis:
- Antibodies against platelet membrane glycoproteins IIb/IIIa or Ib/IX complexes detected in ~80%
- Spleen = major site of antiplatelet antibody production + IgG-coated platelet destruction
- Bone marrow: increased megakaryocytes (compensatory)
Clinical: Insidious onset - petechiae, easy bruising, epistaxis, gum bleeding. Intracerebral hemorrhage is rare but major hazard.
Treatment: Immunosuppressive agents; splenectomy induces complete remission in >2/3 of patients
Heparin-Induced Thrombocytopenia (HIT)
- 3-5% of patients on unfractionated heparin after 1-2 weeks
- IgG antibodies bind platelet factor 4 in heparin-dependent fashion → immune complexes → platelet Fc receptor binding → platelet activation → paradoxical THROMBOSIS (not just bleeding)
- Both venous and arterial thromboses occur
- Treatment: Stop heparin immediately
Thrombotic Thrombocytopenic Purpura (TTP)
Classic pentad: Fever + Thrombocytopenia + Microangiopathic hemolytic anemia + Transient neurologic deficits + Renal failure
Distinguished from HUS (which lacks neurologic symptoms and dominantly presents with acute renal failure in children).
Robbins & Kumar Basic Pathology, p.428-430
7. DIC (Disseminated Intravascular Coagulation)
Definition: Systemic activation of coagulation → fibrin thrombi throughout the microcirculation → consumption of platelets + coagulation factors → secondary fibrinolysis activation. Result: simultaneous clotting AND bleeding in the same patient.
Also called: "Consumptive coagulopathy"
Pathogenesis:
Two main triggers:
- Release of tissue factor / procoagulants into circulation (e.g., placenta in obstetric complications, cancer cells from acute promyelocytic leukemia or adenocarcinoma)
- Widespread endothelial cell damage (bacterial sepsis: endotoxins stimulate tissue factor on monocytes; monocytes release IL-1 and TNF → stimulate tissue factor on endothelium + downregulate thrombomodulin → more thrombin + less protein C activation)
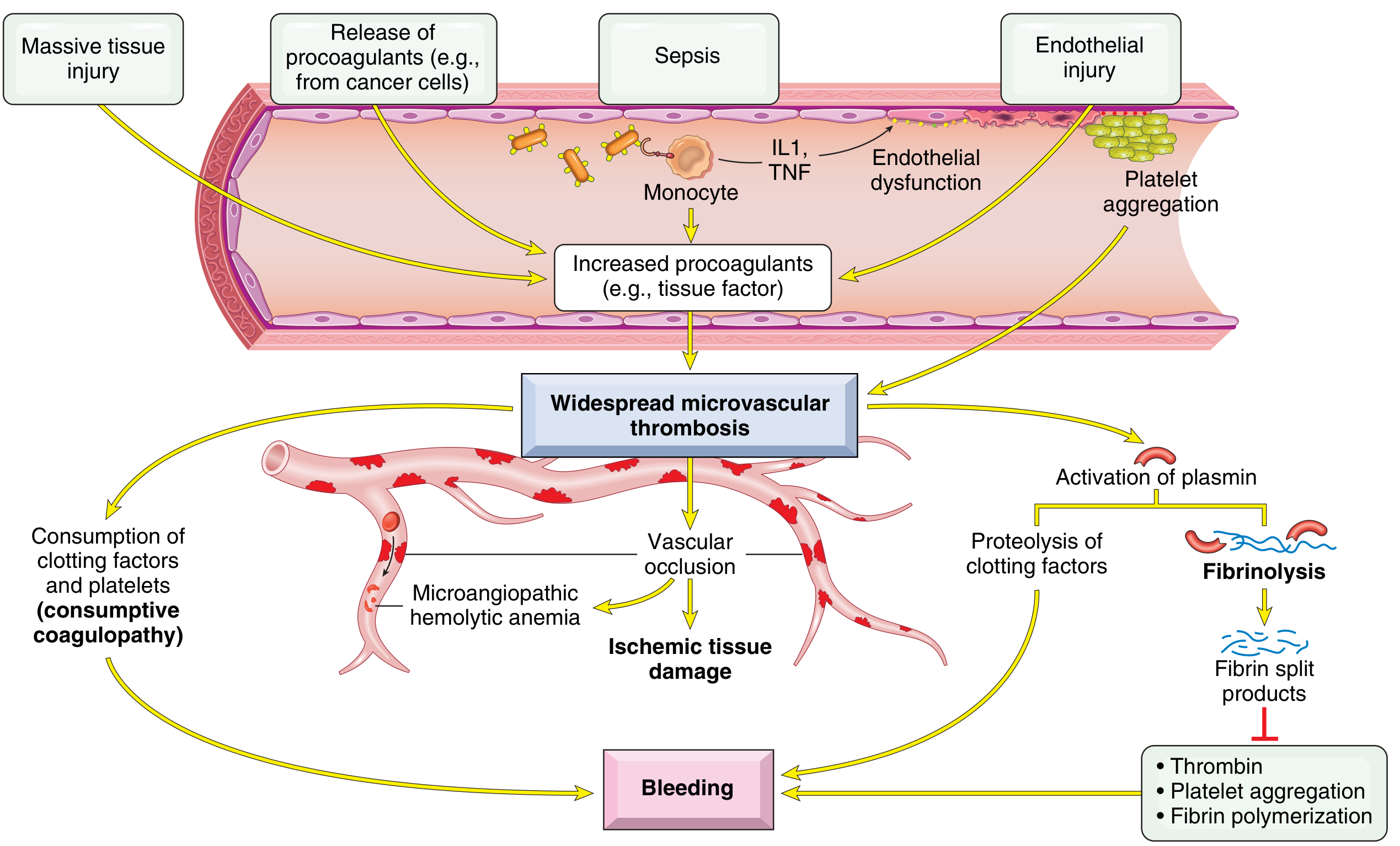
Important causes of DIC:
- Obstetric complications (amniotic fluid embolism, abruptio placentae, retained dead fetus)
- Bacterial sepsis (most common infectious cause)
- Malignancy (acute promyelocytic leukemia, adenocarcinomas)
- Massive tissue injury (trauma, burns, heat stroke)
- Antigen-antibody complex deposition (SLE)
- Infections (meningococci, rickettsiae)
Two Consequences:
- Microvascular thrombi → tissue ischemia → microinfarcts + microangiopathic hemolytic anemia
- Bleeding tendency from consumption of platelets + clotting factors + secondary fibrinolysis (plasmin cleaves fibrin, factor V, factor VIII; fibrin degradation products inhibit platelet aggregation + thrombin activity)
Morphology:
- Microthrombi in kidneys (most severely affected - bilateral cortical necrosis), adrenals (Waterhouse-Friderichsen syndrome), brain, heart
- Diffuse petechiae and ecchymoses on skin, serosal linings, epicardium, endocardium, lungs, urinary mucosa
Clinical:
- Acute DIC: fulminant, dominated by bleeding (e.g., obstetric complications)
- Chronic DIC: dominated by thrombosis (e.g., adenocarcinoma)
- Manifestations: petechiae/ecchymoses, shock, acute renal failure, dyspnea, cyanosis, convulsions, coma
Lab findings:
- Thrombocytopenia
- Prolonged PT and PTT
- Low fibrinogen
- Elevated fibrin degradation products (D-dimer)
Robbins & Kumar Basic Pathology, p.427-428
8. BLOOD GROUPING (ABO System)
Basis of ABO antigens:
ABO antigens are carbohydrates linked to cell surface proteins and lipids, synthesized by polymorphic glycosyltransferases. Present on RBCs, endothelial cells, and some epithelial cells.
Gene located on chromosome 9 encodes a glycosyltransferase with 3 alleles:
- O allele: enzymatically inactive → only H antigen expressed
- A allele: adds N-acetylgalactosamine to H antigen → A antigen
- B allele: adds galactose to H antigen → B antigen
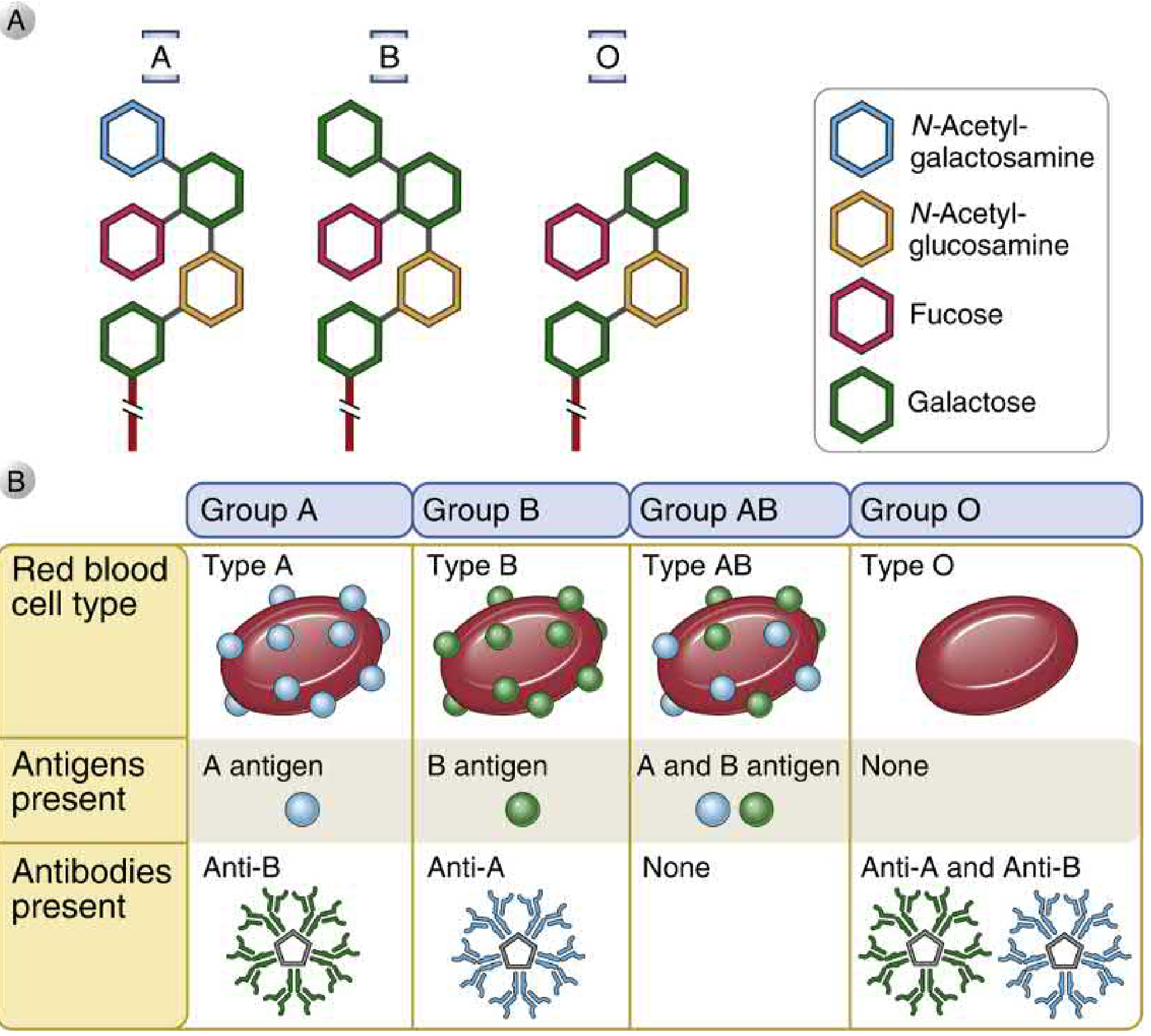
ABO Blood Group Table:
| Blood Type | Genotype | RBC Antigen | Serum Antibody | Universal? |
|---|---|---|---|---|
| A | AA or AO | A antigen | Anti-B | Donor to A, AB |
| B | BB or BO | B antigen | Anti-A | Donor to B, AB |
| AB | AB | A and B antigens | None | Universal recipient |
| O | OO | Neither (only H) | Anti-A and Anti-B | Universal donor |
Why do we have natural antibodies?
Individuals who lack a particular antigen naturally develop IgM antibodies against it - likely from cross-reaction with glycolipids of intestinal bacteria that share structural similarity with ABO antigens.
Bombay Blood Group (Oh):
- Rare mutation in fucosyltransferase gene → cannot produce H antigen
- Cannot produce A, B, or H antigens
- Makes anti-H, anti-A, anti-B antibodies - can only receive blood from other Bombay donors!
Rh System (brief):
- Rh-positive individuals express RhD antigen
- Rh-negative individuals develop anti-D antibodies only after exposure (transfusion or pregnancy)
- Clinically important in haemolytic disease of the newborn (Rh-negative mother + Rh-positive fetus)
Cellular and Molecular Immunology, Robbins, p.1157-1159
Quick Exam Memory Aids
| Topic | Key Buzzword |
|---|---|
| Sickle Cell | Val→Glu at β-globin position 6; deoxygenation triggers sickling |
| Thalassemia | Deficient globin synthesis + intracellular precipitates |
| CML | t(9;22) BCR-ABL Philadelphia chromosome |
| CLL | BCL2 overexpression; BTK signaling; proliferation centers |
| ALL | Most common childhood cancer; hyperploidy = good prognosis |
| Hodgkin | Reed-Sternberg (owl-eye) cells; CD15+, CD30+; from germinal center B cells |
| ITP | Anti-GPIIb/IIIa antibodies; spleen is key; splenectomy cures >2/3 |
| DIC | Consumptive coagulopathy; simultaneous clotting + bleeding |
| HIT | Heparin + anti-PF4 IgG → paradoxical thrombosis |
| ABO | Chromosome 9; carbohydrate antigens; natural IgM antibodies |
Good luck on your exam tomorrow! This guide covers everything from Robbins - focus especially on the pathogenesis mechanisms and the distinguishing features of each condition, as those are typical exam questions.
This is a shared conversation. Sign in to Orris to start your own chat.