13yr old after swimming in a pool presented with vomitings on the same day with fever with bulbar palsy with opthalmoplegia with ptosis on ventilator with normal sensorium
Reading File
Finding Sources
Finding Sources
Finding Sources
Searching PubMed
"Naegleria fowleri" meningoencephalitis treatment
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
I now have comprehensive data to give a thorough, well-structured answer. Here is the full clinical analysis:
Clinical Analysis: 13-year-old with Fever, Vomiting, Bulbar Palsy, Ophthalmoplegia, Ptosis after Swimming
Most Likely Diagnosis: Primary Amoebic Meningoencephalitis (PAM)
Causative organism: Naegleria fowleri
Why PAM Fits This Presentation
| Feature | This Patient | PAM |
|---|---|---|
| Age | 13 years | Median age 14 yrs (range 1 month–85 yrs) |
| Exposure | Swimming pool | Swimming in warm freshwater/poorly maintained pools |
| Onset | Same day | 2–15 day incubation (can be rapid) |
| Fever | Present | Universal — high fever |
| Vomiting | Present | Nausea and vomiting — cardinal feature |
| Cranial nerve palsies | CN III/IV/VI (ptosis + ophthalmoplegia) + bulbar | CN III, IV, VI palsies are common |
| Sensorium | Normal (early) | Initially preserved; progresses to coma |
| Ventilator-dependent | Present | Rapid respiratory compromise requiring ventilation |
"After an incubation period of 2–15 days, severe headache, high fever, nausea, vomiting, and meningismus develop. Photophobia and palsies of the third, fourth, and sixth cranial nerves are common. Rapid progression to seizures and coma may follow. The prognosis is uniformly poor: most patients die within a week." — Harrison's Principles of Internal Medicine 22E, 2025
N. fowleri enters via the olfactory neuroepithelium following nasal inhalation of contaminated water, migrates along the olfactory nerve to the brain, bypasses the blood-brain barrier, and invokes a fulminant neutrophilic meningoencephalitis. Even a poorly maintained swimming pool is a recognized source — the CDC has documented PAM from chlorinated pools.
Differential Diagnosis
1. Botulism (important to consider — normal sensorium + cranial palsies)
- Bilateral cranial nerve palsies (ptosis, ophthalmoplegia, bulbar = dysarthria/dysphagia) + descending flaccid paralysis + fully conscious patient is the classic botulism triad
- Against it: Fever is NOT a feature of botulism; CSF is normal; no GI prodrome of toxin-type (unless wound/food)
- Normal CSF protein distinguishes botulism from GBS
2. Miller Fisher Syndrome (GBS variant)
- Triad: ophthalmoplegia + ataxia + areflexia
- Can have bulbar weakness; can require ventilation
- Against it: Fever is atypical; swimming pool context not relevant; usually follows a viral infection 1–3 weeks prior; CSF shows albuminocytologic dissociation (elevated protein, no cells)
3. Brainstem Encephalitis (Rhombencephalitis)
- Listeria monocytogenes, Enterovirus 71, HSV-2 can cause brainstem involvement with cranial nerve palsies + fever
- Consider: Enterovirus rhombencephalitis in children
4. Bacterial Meningitis with Cranial Nerve Involvement
- CN VI palsy is most common in bacterial meningitis (elevated ICP)
- Combined ophthalmoplegia + bulbar + fever: consider TB meningitis (basal meningitis)
5. Wernicke's Encephalopathy — ophthalmoplegia + ataxia, but no fever; unlikely in 13-year-old
Why the Sensorium is Normal (Key Feature)
In early PAM, sensorium is preserved. The brain-eating amoeba first produces a fulminant meningitic picture with cranial nerve palsies before cerebral involvement causes drowsiness and coma. This is also characteristic of botulism (fully conscious throughout). This preserved sensorium with cranial nerve deficits and fever most strongly points to PAM in this swimming pool context.
Investigations
| Test | Expected Finding in PAM |
|---|---|
| CSF wet mount | Motile trophozoites — diagnostic |
| CSF cell count | High WBC (up to 20,000/μL) — neutrophilic |
| CSF protein | Elevated |
| CSF glucose | Low |
| CSF Gram stain/culture | Negative for bacteria |
| CSF PCR (CDC) | Positive for N. fowleri |
| Brain MRI | Diffuse enhancement, particularly olfactory bulbs, basal ganglia |
| Serology | Not useful (antibodies found in healthy adults) |
Critical point: CSF should be examined immediately as a fresh wet preparation — trophozoites (10–35 μm) are motile and can be seen on direct microscopy. Refrigeration kills them. Contact CDC Emergency Operations at (770) 488-7100 immediately.
Treatment (Medical Emergency)
PAM has a >97% fatality rate, but survivors have been reported with early aggressive therapy:
Combination Regimen (All survivors included these agents):
| Drug | Role |
|---|---|
| Miltefosine (Impavido) | Most critical — only drug shown in all 7 worldwide survivors; alkylphosphocholine amoebicidal |
| Amphotericin B liposomal (IV + intrathecal) | Primary amoebicidal |
| Fluconazole | Synergistic |
| Azithromycin | Synergistic |
| Rifampin | Synergistic |
| Dexamethasone | Reduce neuroinflammation |
Adjunctive:
- Therapeutic hypothermia — associated with better outcomes in survivors
- Aggressive ICP management
- Mechanical ventilation (already on)
"All seven survivors among reported worldwide cases (381) included miltefosine as well as the five drugs above. Early diagnosis, prompt combination therapy including miltefosine, and aggressive management of neurologic complications, including therapeutic hypothermia, are important factors in better outcomes." — Harrison's 22E
Summary Algorithm
13yo + fever + vomiting + swimming pool + CN III/IV/VI palsies + bulbar palsy + ventilator + NORMAL SENSORIUM
│
↓
PRIMARY AMOEBIC MENINGOENCEPHALITIS (Naegleria fowleri) — MOST LIKELY
│
↓
URGENT: Fresh CSF wet mount for motile trophozoites
+ CDC PCR (call CDC immediately)
│
↓
START EMPIRIC TREATMENT WITHOUT WAITING:
Miltefosine + Liposomal AmB + Fluconazole + Azithromycin + Rifampin + Dexamethasone
+ Therapeutic hypothermia
Prognosis
Prognosis is grave — mortality >97% even with treatment. Only 7 survivors have been documented worldwide (out of 381 reported global cases as of 2022). Survival correlates with:
- Early suspicion and diagnosis
- Prompt initiation of miltefosine-containing combination therapy
- Aggressive ICU/neurological management
Recent PubMed evidence (Hall AD et al., 2024, PMID 38182931 — review of successfully treated cases) confirms miltefosine as central to the rare survivors.
Sources: Harrison's Principles of Internal Medicine 22E (2025); Bradley & Daroff's Neurology in Clinical Practice; Medical Microbiology 9e; Tintinalli's Emergency Medicine; Red Book 2021
Without meningismus
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
The absence of meningismus is a pivotal negative that fundamentally redirects the diagnosis. Here is the revised analysis:
Revised Diagnosis: No Meningismus Changes Everything
The full syndrome now is:
Fever + Vomiting + Bulbar palsy + Ophthalmoplegia + Ptosis + Ventilator-dependent + Normal sensorium + NO neck stiffness/meningism
This combination — cranial nerve palsies descending to bulbar involvement + respiratory failure + intact consciousness + NO meningeal signs — is the hallmark of a neuromuscular junction (NMJ) disorder, most specifically:
#1 Diagnosis: Wound/Water-Associated Botulism
Why This Is Now the Leading Diagnosis
| Feature | This Patient | Botulism |
|---|---|---|
| Ptosis | ✅ | Classic — first cranial nerve sign |
| Ophthalmoplegia (CN III/IV/VI) | ✅ | Characteristic bilateral cranial palsies |
| Bulbar palsy (dysarthria, dysphagia) | ✅ | Descending cranial → bulbar pattern |
| Normal sensorium (fully awake) | ✅ | Brain is unaffected — pathognomonic |
| Ventilator required | ✅ | Respiratory muscle paralysis |
| No meningismus | ✅ | No CNS inflammation whatsoever |
| Vomiting | ✅ | Autonomic involvement; also foodborne prodrome |
| Swimming pool | ✅ | Wound botulism from water/soil contact |
"The distinct syndrome of bilateral cranial palsies and descending flaccid paralysis in a fully conscious patient should render the diagnosis of botulism straightforward." — Harrison's Principles of Internal Medicine 22E
"No fever or other signs of infection occur [in classic botulism]. A slower moving form of the disease occurs when the toxin is produced endogenously in the intestinal tract or a wound." — Sherris & Ryan's Medical Microbiology 8e
The Fever — How Does It Fit?
Fever is not typical of foodborne botulism (preformed toxin). However, wound botulism — where C. botulinum infects a wound (or skin abrasion from a pool/lake floor) and produces toxin in situ — CAN produce fever due to the wound infection itself. A 13-year-old swimming could sustain minor abrasions, and pool environments can be contaminated with C. botulinum spores from soil.
Wound botulism with secondary wound infection = fever + all neurological features + no meningismus.
Pathophysiology of the Neurological Pattern
C. botulinum toxin (metalloproteinase)
↓
Cleaves SNARE proteins at presynaptic membrane
↓
Blocks ACh release at NMJ
↓
Descending flaccid paralysis:
CRANIAL NERVES FIRST (CN III/IV/VI → ptosis + ophthalmoplegia)
↓
BULBAR (CN IX/X/XII → dysphagia, dysarthria, dysphonia)
↓
RESPIRATORY MUSCLES (diaphragm + intercostals → ventilator)
↓
LIMB WEAKNESS (late)
*** Brain unaffected → Normal sensorium throughout ***
*** No meningeal inflammation → No neck stiffness ***
Differential Diagnosis (with No Meningismus)
1. Wound Botulism ← Most Likely
- Swimming pool abrasion → C. botulinum spore germination → in situ toxin → descending NMJ blockade
- Fever from wound infection
- Normal CSF
2. Miller Fisher Syndrome (GBS variant)
- Triad: ophthalmoplegia + ataxia + areflexia
- No fever typically; can have bulbar; CSF shows elevated protein (albuminocytologic dissociation)
- Anti-GQ1b antibodies positive
- Against it: Fever is atypical; can have some overlap but no meningismus fits; ataxia is the key feature (check for it)
3. Foodborne Botulism
- If the child consumed contaminated food around the time of swimming
- Vomiting fits the gastrointestinal prodrome (nausea, vomiting, cramping precede neurological symptoms by hours)
- No fever expected — but possible co-existing illness
4. Myasthenia Gravis (MG) Crisis
- Ptosis + ophthalmoplegia + bulbar + respiratory failure
- Against: Fever not a feature; no exposure history; fatigability worse with exertion; usually chronic history; Tensilon test positive
5. Bickerstaff Brainstem Encephalitis
- Ophthalmoplegia + ataxia + hyperreflexia + impaired consciousness
- Against: Sensorium should be impaired; no fever typically
6. Enterovirus 71 Rhombencephalitis
- Children, fever, brainstem involvement, respiratory failure
- But: usually has altered sensorium, myoclonus, autonomic instability; typically preceded by hand-foot-mouth disease
Investigations
| Test | Expected in Wound Botulism |
|---|---|
| Serum botulinum toxin assay (mouse bioassay) | Positive — gold standard |
| Wound culture | C. botulinum growth under anaerobic conditions |
| CSF | Normal — protein, glucose, cells all normal |
| EMG/NCS | Facilitating pattern on rapid repetitive stimulation (incremental response), reduced CMAP amplitude |
| CT/MRI brain | Normal |
| Acetylcholine receptor antibodies | Negative (rules out MG) |
| Anti-GQ1b antibodies | Negative (rules out MFS) |
| Edrophonium (Tensilon) test | Negative in botulism |
Key CSF distinction:
- Botulism → Normal CSF
- GBS/MFS → Elevated protein, no cells
- PAM → Neutrophilic pleocytosis, low glucose
- Bacterial meningitis → Neutrophilic pleocytosis + positive culture
Treatment
Wound Botulism:
- Heptavalent Botulinum Antitoxin (HBAT) — give immediately, before lab confirmation; binds circulating free toxin (cannot reverse bound toxin)
- Wound debridement + Penicillin G or Metronidazole (to eliminate C. botulinum from wound)
- Mechanical ventilation — may be required for weeks to months
- Supportive ICU care — recovery depends on new NMJ sprouting (weeks to months)
- Do NOT give aminoglycosides — worsen NMJ blockade
- Contact CDC (Emergency Operations: 770-488-7100) for antitoxin release
Summary
Without meningismus, PAM drops significantly in likelihood (it characteristically presents like bacterial meningitis with meningeal signs). The clinical triad of:
Descending cranial nerve palsies (ptosis → ophthalmoplegia → bulbar) + respiratory failure requiring ventilation + fully conscious patient + NO meningism
...is the signature of botulism until proven otherwise — specifically wound botulism in the context of swimming pool exposure with fever.
The normal sensorium with no meningismus is the single most important negative that points the diagnosis away from meningitis/encephalitis and squarely toward a presynaptic NMJ toxin-mediated disorder.
First is bulbar
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Searching PubMed
"pharyngeal-cervical-brachial" variant Guillain-Barre syndrome
Reading File
Reading Article
PMID: 25072194
Now the picture is crystal clear. Here is the revised, refined analysis:
Revised Analysis: Bulbar Palsy First, Then Ophthalmoplegia + Ptosis
The Sequence Is the Diagnosis
Bulbar (CN IX/X/XII) FIRST
↓
Ophthalmoplegia + Ptosis (CN III/IV/VI) NEXT
↓
Respiratory failure (ventilator)
↓
Normal sensorium | No meningismus | Fever | Post-swimming
This ascending cranial nerve pattern — starting low (bulbar) and moving up (ocular) — is the inverse of botulism and points squarely to:
#1 Diagnosis: Miller Fisher Syndrome / GBS Overlap (Pharyngeal-Cervical-Brachial variant or MFS-GBS overlap)
Specifically: GBS with severe bulbar and facial/cranial nerve paralysis, or the MFS-GBS overlap syndrome
Why
Harrison's 22E explicitly identifies this GBS subtype:
"GBS with severe bulbar and facial paralysis, sometimes associated with antecedent cytomegalovirus (CMV) infection and anti-GM2 antibodies" — as a regional GBS variant
And the Wakerley/Yuki 2014 classification (Nature Reviews Neurology, PMID 25072194) defines:
- Pharyngeal-cervical-brachial (PCB) weakness — a localized GBS form presenting with oropharyngeal, neck, and shoulder weakness (bulbar first), which can extend to involve ocular muscles
- MFS-GBS overlap — begins with ophthalmoplegia/ataxia then develops limb/bulbar weakness
- Bickerstaff Brainstem Encephalitis (BBE) — ophthalmoplegia + ataxia + impaired consciousness (against this here as sensorium is normal)
The PCB variant is particularly compelling:
- Starts with dysphagia, dysarthria (bulbar = CN IX/X/XII)
- Progresses upward to involve CN III/IV/VI (ophthalmoplegia, ptosis)
- Can progress to respiratory failure requiring ventilation
- Normal sensorium throughout
- Associated with anti-GT1a antibodies (not GQ1b, which is MFS)
Differential Framework by Cranial Nerve Sequence
| Pattern | Direction | Diagnosis |
|---|---|---|
| CN III/IV/VI → Bulbar → Limbs | Descending | Botulism |
| Bulbar → CN III/IV/VI → Limbs | Ascending / caudal→rostral | PCB-GBS, MFS-GBS overlap |
| Bulbar + CN III/IV/VI simultaneously | Diffuse brainstem | Brainstem encephalitis (BBE, rhombencephalitis) |
| Bulbar + CN III/IV/VI + altered consciousness | Brainstem | Bickerstaff's |
Full Differential (Reordered by Fit)
1. Pharyngeal-Cervical-Brachial (PCB) variant of GBS ← Top Diagnosis
- Bulbar weakness first (dysphagia, dysarthria, nasal voice)
- Extends to CN III/IV/VI (ophthalmoplegia, ptosis)
- Respiratory failure in severe cases
- Normal sensorium, no meningismus
- CSF: albuminocytologic dissociation (elevated protein, no cells)
- Anti-GT1a IgG antibodies — highly specific for PCB
- Trigger: swimming pool → possible enteric infection (Campylobacter, CMV) 1–3 weeks prior
2. MFS-GBS Overlap Syndrome
- Classic MFS (ophthalmoplegia + ataxia + areflexia) with additional bulbar/limb weakness
- Can begin with bulbar if overlap is significant
- Anti-GQ1b IgG (present in >85% MFS cases)
- Fever uncommon but possible with antecedent infection
3. Bickerstaff Brainstem Encephalitis (BBE)
- Ophthalmoplegia + ataxia + impaired/fluctuating consciousness
- Anti-GQ1b positive (same spectrum as MFS)
- Against here: sensorium is described as normal
- MRI may show brainstem hyperintensities
4. Botulism (Wound)
- Now less likely given bulbar-first sequence (botulism is strictly cranial-first descending: CN III/IV/VI → bulbar)
- However, wound botulism with a scratch from pool floor remains possible if one argues the sequence was misidentified
5. Listeria Rhombencephalitis
- Fever + brainstem involvement (CN palsies + bulbar)
- Usually has altered sensorium or cerebellar signs
- CSF typically abnormal
- Against: normal sensorium, no meningismus
Why Swimming Pool is Still Relevant
The pool provides the antecedent infection trigger for an immune-mediated neuropathy (GBS/PCB/MFS):
- Pool water → swallowing of pool water → enteric infection (Campylobacter jejuni, CMV, hepatitis E)
- Incubation 1–3 weeks → molecular mimicry → anti-ganglioside antibodies → cranial nerve demyelination
- The same-day onset suggests either a very rapid post-infectious response or the swimming event itself is coincidental while the antibody-mediated attack was already underway
Investigations
| Test | Expected Finding |
|---|---|
| CSF | Elevated protein, NO pleocytosis (albuminocytologic dissociation) |
| Anti-GT1a IgG | Positive → confirms PCB variant |
| Anti-GQ1b IgG | Positive → confirms MFS/BBE |
| Anti-GM2 IgG | Positive → GBS with bulbar/cranial involvement post-CMV |
| Nerve conduction studies | Demyelinating pattern (prolonged distal latencies, reduced conduction velocity) or axonal |
| MRI brain/brainstem | Normal (PCB/MFS) or brainstem T2 hyperintensity (BBE) |
| Stool/serology | Campylobacter jejuni, CMV, EBV |
| EMG | Reduced CMAP, absent/reduced F waves |
Treatment
Since the likely diagnosis is immune-mediated (GBS spectrum):
- IVIg (Intravenous immunoglobulin) — 2 g/kg over 5 days — first line
- Plasmapheresis — equally effective alternative; 4–6 exchanges
- Do NOT use steroids — not beneficial in GBS and may worsen
- Mechanical ventilation — already established; monitor FVC, NIF
- Supportive care: DVT prophylaxis, nutrition, dysautonomia monitoring, pain management
Key Teaching Point
Bulbar-first then ophthalmoplegia = ascending cranial nerve pattern = PCB-GBS or MFS-GBS overlapOphthalmoplegia-first then bulbar = descending pattern = BotulismThe direction of cranial nerve involvement is the single most important localizing clue in this presentation.
The swimming pool provides the antigenic trigger (enteric infection) for an immune-mediated post-infectious cranial polyneuropathy — not direct CNS invasion as in PAM, which has been effectively excluded by the absence of meningismus and the ascending cranial nerve sequence.
Palatal tremors vs palatal myoclonus
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Excellent, comprehensive data from three major neurology textbooks. Here is the full comparison:
Palatal Tremor vs Palatal Myoclonus
Terminology First
These two terms refer to the same physical phenomenon (rhythmic palatal movements) but from different conceptual frameworks:
- "Palatal myoclonus" — older term; used because the movements appeared myoclonic
- "Palatal tremor" — current preferred term; reclassified because of persistent rhythmicity (tremors are rhythmic; myoclonus is typically arrhythmic)
However, the distinction matters mechanistically:
- True tremor = oscillatory movement from synchronous or alternating contractions of antagonist muscles
- True myoclonus = rhythmic contractions of agonist muscles only (uniphasic)
Palatal movements are agonist-only contractions → technically myoclonus by mechanism, but tremor by pattern. Both terms remain in use.
The Two Subtypes: The Critical Clinical Distinction
| Feature | Essential Palatal Tremor (EPT) | Symptomatic Palatal Tremor (SPT) |
|---|---|---|
| Old name | Essential palatal myoclonus | Symptomatic palatal myoclonus |
| Muscle involved | Tensor veli palatini (CN V₃) | Levator veli palatini (CN X) |
| Ear click | ✅ Present — characteristic (opens Eustachian tube) | ❌ Absent |
| Persists during sleep | ❌ Disappears with sleep | ✅ Persists during sleep |
| Associated movements | Palate only | Palate + pharynx + face + eyes + diaphragm + neck/shoulders |
| Pendular nystagmus | ❌ Absent | ✅ May be present (synchronized) |
| MRI brain | Normal — no lesion | Inferior olivary pseudohypertrophy (hallmark) |
| Brainstem lesion | None | ✅ Central tegmental tract lesion |
| Guillain-Mollaret triangle | Not involved | ✅ Disrupted — key pathology |
| Etiology | Unknown / functional | Vascular, demyelinating, neoplastic, traumatic |
| Frequency | Variable; often higher | Variable but tends to be fixed |
| Functional/psychogenic | More likely | Less likely |
| Onset after injury | — | Months of latency before tremor appears |
Pathoanatomy of Symptomatic Palatal Tremor
The key structure is the Guillain-Mollaret Triangle (dentato-rubro-olivary pathway):
Contralateral Dentate Nucleus (cerebellum)
↓ (superior cerebellar peduncle / brachium conjunctivum)
Ipsilateral Red Nucleus (midbrain)
↓ (Central Tegmental Tract — CTT)
Ipsilateral Inferior Olivary Nucleus (medulla)
↓ (inferior cerebellar peduncle / climbing fibers)
Back to Dentate Nucleus
Lesion of the CTT (or anywhere in the triangle) → disinhibits the inferior olivary nucleus → olive undergoes pseudohypertrophy (hypertrophic olivary degeneration, HOD) → rhythmic olivary discharges → palatal tremor
With unilateral palatal tremor → it is the contralateral olive that enlarges (because the CTT crosses: lesion on one side → contralateral olive disinhibited)
MRI in SPT: Hypertrophic Olivary Degeneration (HOD)
- T2/FLAIR: hyperintensity in inferior olive
- Enlargement of olive (not atrophy — hence "hypertrophy")
- Appears months after the causative lesion
- May eventually regress over years
Quick Comparison Summary
| Essential (EPT) | Symptomatic (SPT) | |
|---|---|---|
| Ear click | ✅ Yes | ❌ No |
| Sleep | Disappears | Persists |
| Muscle | Tensor veli palatini | Levator veli palatini |
| MRI | Normal | Olivary pseudohypertrophy |
| Cause | Unknown | Guillain-Mollaret triangle lesion |
| Other movements | No | Yes (eyes, face, diaphragm) |
Associated Syndromes of SPT
-
Oculopalatal myoclonus
- Lateral form: jerky nystagmoid eye movements + oblique/rotatory components + lateralized palatal myoclonus (unilateral lesion)
- Midline form: vertical pendular eye movements + bilateral symmetric palatal myoclonus
-
Progressive Ataxia and Palatal Tremor (PAPT)
- Low-frequency palatal tremor + progressive cerebellar degeneration + vertical nystagmus + internuclear ophthalmoplegia
- Associated with Alexander disease, mitochondrial disorders, POLG mutations
- Familial PAPT: brainstem/cervical cord atrophy, NO olivary pseudohypertrophy
-
Whipple disease — oculomasticatory myorhythmia (masseter contractions + pendular ocular convergence; analogous phenomenon)
Treatment
| Agent | Use |
|---|---|
| Gabapentin | Best evidence; "dramatic effect" in some cases |
| Clonazepam | 0.25–0.5 mg → up to 6 mg/day |
| Sodium valproate | 250 mg → up to 1000 mg/day |
| Tetrabenazine / Haloperidol | Occasionally helpful |
| Botulinum toxin (local injection) | Modest relief; best for ear click in EPT |
"Selective injection of the palatal muscles with botulinum toxin, while technically demanding, affords modest relief; it is particularly helpful in eliminating the bothersome ear clicking." — Adams & Victor's Neurology 12e
Memory Hook
Essential = Ear click, Extinguished in sleep, Extra-normal MRI, Etiologically unknownSymptomatic = Sleep-persistent, Synchronized with nystagmus, Striking olivary hypertrophy, Secondary to Guillain-Mollaret triangle lesion
— Adams & Victor's Principles of Neurology 12e; Bradley & Daroff's Neurology in Clinical Practice; Localization in Clinical Neurology 8e
Rhombencephalitis
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
I now have all the data needed for a comprehensive answer.
Rhombencephalitis
Definition
Rhombencephalitis = inflammation of the rhombencephalon (hindbrain) — encompassing the brainstem (pons + medulla) and cerebellum. The term is used when encephalitis is anatomically restricted to these posterior fossa structures, as opposed to diffuse encephalitis affecting the whole brain.
The rhombencephalon is so named because the embryological fourth ventricle has a rhomboid (diamond) shape in dorsal view.
Clinical Syndrome
The clinical features reflect the anatomy — all structures of the posterior fossa:
| Structure Involved | Clinical Features |
|---|---|
| Pons | CN V (facial sensory loss), CN VI (lateral gaze palsy), CN VII (facial palsy), CN VIII (hearing, vertigo), long tract signs |
| Medulla | CN IX/X (bulbar palsy — dysphagia, dysarthria, dysphonia), CN XII (tongue weakness), respiratory failure |
| Cerebellum | Ataxia (gait, limb), dysmetria, nystagmus |
| Midbrain (if involved) | CN III/IV palsy, altered consciousness |
| General brainstem | Diplopia, internuclear ophthalmoplegia, autonomic instability |
Characteristic Biphasic Course (especially Listeria)
- Phase 1 (days 1–5): Fever, headache, nausea, vomiting — prodromal
- Phase 2 (days 5–10): Asymmetric cranial nerve palsies, cerebellar signs, long-tract signs, respiratory failure
Causes — Classified
Infectious
Bacterial
| Organism | Notes |
|---|---|
| Listeria monocytogenes | Classic cause — "rhombencephalitis" is almost synonymous with Listeria; predilects brainstem; asymmetric cranial nerve palsies; CSF often mildly abnormal or even normal; MRI shows brainstem T2 signal; meningeal signs in only 50% |
| Burkholderia pseudomallei | Melioidosis — similar to Listeria; endemic in India/Southeast Asia; multiple small abscesses in cerebellum and white matter |
| Mycobacterium tuberculosis | Basal meningitis → brainstem involvement by exudate; cranial nerve palsies common |
| Brucella | Neurobrucellosis; chronic form |
| Treponema pallidum | Neurosyphilis |
Viral
| Virus | Notes |
|---|---|
| Enterovirus 71 (EV71) | Children; follows hand-foot-mouth disease; brainstem encephalitis with myoclonic jerks + limb trembling; MRI: brainstem lesions; can progress to pulmonary edema/hemorrhage; most deaths in children ≤5 years |
| HSV-1 / HSV-2 | Usually temporal lobe but brainstem forms occur; recurrent brainstem encephalitis described with HSV-2 |
| EBV, CMV | Post-infectious; immunocompromised |
| Rabies | Brainstem + spinal cord syndromes |
| Tick-borne encephalitis virus | Brainstem/cerebellar involvement |
| West Nile Virus | Brainstem encephalitis, flaccid paralysis |
| SARS-CoV-2 | Rare brainstem involvement |
Fungal / Parasitic
| Organism | Notes |
|---|---|
| Cryptococcus | Basilar meningitis → brainstem |
| Aspergillus | Angioinvasive; immunocompromised |
Autoimmune / Para-infectious
| Entity | Key Features |
|---|---|
| Bickerstaff Brainstem Encephalitis (BBE) | Ophthalmoplegia + ataxia + impaired consciousness (distinguishes from MFS); anti-GQ1b antibodies; MRI: T2 brainstem/thalamic hyperintensity or normal; treat as GBS spectrum (IVIg/plasma exchange) |
| CLIPPERS | Chronic Lymphocytic Inflammation with Pontine Perivascular Enhancement Responsive to Steroids; subacute ataxia, diplopia, dysarthria; MRI: punctate gadolinium enhancement in pons/cerebellum; responds dramatically to steroids |
| Anti-NMDAR encephalitis | Usually limbic but brainstem forms |
| Neurosarcoidosis | Cranial nerve palsies, hypothalamic involvement |
| CNS vasculitis | |
| Multiple sclerosis | Brainstem demyelinating plaques |
| Paraneoplastic | Anti-Hu, anti-Ri (brainstem/cerebellar) |
Key Organism Comparison: Listeria vs Enterovirus 71
| Feature | Listeria Rhombencephalitis | EV71 Rhombencephalitis |
|---|---|---|
| Age | Elderly, immunocompromised, neonates | Young children (≤5 years) |
| Preceding illness | None / subtle GI | HFMD (hand-foot-mouth disease) |
| Onset pattern | Biphasic — prodrome then CN palsies | Acute |
| CN palsies | Asymmetric, multiple | Present |
| Myoclonus/tremor | Less typical | ✅ Characteristic — myoclonic jerks, limb trembling |
| Autonomic storm | Less prominent | ✅ Common — tachycardia, hypertension |
| CSF | Mild pleocytosis or normal | Mild lymphocytic pleocytosis |
| MRI | T2 brainstem lesions | Brainstem, dorsal pons/medulla lesions |
| Blood cultures | Positive in >75% | Negative |
| Treatment | Ampicillin + Gentamicin | Supportive |
| Mortality | ~30% | High in severe cases |
Bickerstaff Brainstem Encephalitis — Special Status
BBE occupies the interface between infectious rhombencephalitis and immune-mediated brainstem disease:
- Preceded by systemic viral infection
- Ophthalmoplegia + ataxia + altered consciousness (consciousness = key differentiator from MFS)
- Anti-GQ1b IgG antibodies — same spectrum as Miller Fisher Syndrome
- MRI: T2 hyperintensity in brainstem, cerebellum, thalamus — or normal
- CSF: elevated protein, no cells (same as GBS/MFS)
- Treatment: IVIg or plasmapheresis (as for GBS)
- BBE + limb weakness = MFS-GBS overlap
"A brainstem disorder often confused with infection is Bickerstaff's brainstem encephalitis. Patients with this disorder have often had a preceding systemic viral infection, then acutely develop ataxia, ophthalmoplegia, long-tract signs, and alterations of consciousness including coma." — Plum & Posner's Diagnosis and Treatment of Stupor and Coma
Investigations
| Test | Significance |
|---|---|
| MRI brain (FLAIR/T2/DWI) | T2 hyperintensity in brainstem/cerebellum; always first |
| CSF | Cell count, protein, glucose, culture, PCR (HSV, EV, CMV) |
| Blood cultures × 3 | Positive in >75% of Listeria — do before antibiotics |
| Anti-GQ1b antibodies | BBE / MFS |
| Anti-GT1a antibodies | PCB-GBS variant |
| EV71 PCR | Throat swab, stool, CSF |
| Listeria PCR | CSF or blood |
| Brucella serology | Endemic areas |
Treatment by Etiology
| Cause | Treatment |
|---|---|
| Listeria | Ampicillin 2g IV q4h + Gentamicin (penicillin allergy: TMP-SMX) |
| Burkholderia (Melioidosis) | Ceftazidime or meropenem → maintenance cotrimoxazole |
| HSV | Acyclovir 10mg/kg IV q8h |
| EV71 | Supportive; milrinone for autonomic storm |
| BBE / GBS spectrum | IVIg 2g/kg over 5 days or plasmapheresis |
| CLIPPERS | High-dose corticosteroids → immunosuppression maintenance |
| TB | Anti-tubercular therapy + steroids |
Summary Framework
RHOMBENCEPHALITIS
│
├─ INFECTIOUS
│ ├─ Bacterial: Listeria ★ (classic), Melioidosis, TB, Brucella
│ ├─ Viral: Enterovirus 71 ★ (children), HSV-2, Rabies, Tick-borne
│ └─ Fungal: Cryptococcus, Aspergillus
│
├─ AUTOIMMUNE / PARA-INFECTIOUS
│ ├─ Bickerstaff Brainstem Encephalitis (anti-GQ1b) ★
│ ├─ CLIPPERS (pontine perivascular)
│ ├─ Paraneoplastic (anti-Hu, anti-Ri)
│ └─ MS / neurosarcoidosis
│
└─ EMPIRIC APPROACH (while awaiting results)
├─ Cover Listeria: Ampicillin + Gentamicin
├─ Cover HSV: Acyclovir
└─ Consider IVIg if anti-GQ1b pending (BBE)
Sources: Adams & Victor's Principles of Neurology 12e; Bradley & Daroff's Neurology in Clinical Practice; Harrison's Principles of Internal Medicine 22E; Plum & Posner's Diagnosis and Treatment of Stupor and Coma; Localization in Clinical Neurology 8e
Generalised dystonia in 20yr old from 5 years
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive data. Let me synthesize the full answer.
Generalised Dystonia in a 20-Year-Old (Onset at Age 15)
Clinical Context
- Age of onset: 15 years (adolescent-onset)
- Current age: 20 years
- Duration: 5 years, progressive
- Distribution: Generalised
This profile — adolescent-onset, generalised, progressive over years — places the patient in a specific diagnostic category that differs fundamentally from adult-onset focal dystonias.
Classification Framework (Consensus 2013 — Two-Axis System)
Axis I: Clinical Characteristics
| Feature | This Patient |
|---|---|
| Age at onset | Adolescence (13–20 years) |
| Body distribution | Generalised |
| Temporal pattern | Progressive (5 years) |
| Variability | Persistent |
Axis II: Etiology
| Category | Examples relevant here |
|---|---|
| Inherited | DYT1, DYT2, DYT5, DYT6, DYT17, Wilson disease, PKAN |
| Acquired | Perinatal injury, drugs, infection, vascular, psychogenic |
| Idiopathic | No structural or degenerative pathology found |
Key rule: In a young patient with generalised dystonia, always rule out treatable/secondary causes first before labelling as primary/genetic.
Priority Diagnosis: Wilson Disease ← Must Exclude First
Wilson disease (hepatolenticular degeneration) is the single most important diagnosis to exclude in any young patient with generalised dystonia because:
- It is treatable — delay causes irreversible neurological damage
- It presents exactly in this age group (5–35 years, peak 2nd decade)
- Neurological Wilson's = basal ganglia → generalised dystonia is a cardinal feature
Wilson Disease Features
- Autosomal recessive (ATP7B gene, chromosome 13)
- Copper accumulates in basal ganglia (especially putamen), liver, cornea
- Neurological forms: dystonia (most common), tremor, dysarthria, dysphagia, parkinsonism, psychiatric symptoms
- Kayser-Fleischer rings — golden-brown corneal deposits (slit-lamp examination)
- Investigations: Serum ceruloplasmin ↓, 24-hr urinary copper ↑, serum copper ↓, slit-lamp KF rings, MRI ("face of giant panda" sign in midbrain)
- Treatment: D-penicillamine, trientine, zinc salts — dramatic improvement if early
Differential Diagnosis: Generalised Dystonia, Adolescent Onset
1. Primary (Isolated) Genetic Dystonias
| Gene/DYT | Name | Inheritance | Onset | Features |
|---|---|---|---|---|
| DYT1 / TOR1A | Oppenheim dystonia (Early-onset primary torsion dystonia) | AD (30% penetrance) | 6–12 yrs (can extend to adolescence) | Starts in one foot/leg while walking → spreads to generalised; Chr 9q; Torsin A protein; no cognitive/intellectual involvement |
| DYT2 | AR primary torsion dystonia | AR | Childhood–adolescence | Similar to DYT1; consanguineous families |
| DYT6 / THAP1 | Adolescent-onset mixed torsion dystonia | AD | Adolescence | Upper limb or cranial onset; dysphonia prominent; German-Mennonite origin |
| DYT17 | Adolescent-onset AR torsion dystonia | AR | Adolescence | Rare |
DYT1 is the most common inherited generalised dystonia. It begins in one limb (usually a leg/foot during walking), progresses over years to become generalised. Adolescent onset at 15 is within range.
2. Heredodegenerative Diseases Causing Generalised Dystonia
| Disease | Key Feature | Test |
|---|---|---|
| Wilson disease | Liver + neuro + KF rings | Ceruloplasmin, 24hr urine Cu, MRI |
| PKAN (NBIA type 1) | "Eye of the tiger" MRI sign (GPi); PANK2 mutation; AR | MRI brain (T2 GPi hypointensity with central hyperintensity) |
| Neuroacanthocytosis | Orolingual dystonia, chorea, self-mutilation, areflexia | Blood film (acanthocytes), VPS13A gene |
| Huntington disease (juvenile) | Akinetic-rigid or dystonic form; cognitive decline | CAG repeat ≥55; family history |
| DRPLA | Ataxia + myoclonus + dystonia + dementia | Genetic testing |
| GM1/GM2 gangliosidosis | Cherry-red spot, cognitive decline | Enzyme assay |
| Metachromatic leukodystrophy | White matter disease, MRI periventricular changes | Arylsulfatase A |
| Niemann-Pick C | Vertical supranuclear gaze palsy, ataxia | Filipin staining, NPC1/2 |
| Lesch-Nyhan syndrome | Self-mutilation, hyperuricaemia (males only) | HPRT enzyme activity |
3. Dopa-Responsive Dystonia (DRD) — DYT5 / Segawa Syndrome
Cannot miss this — entirely treatable with levodopa:
- GCH1 gene (autosomal dominant) or TH gene (AR)
- Onset in childhood/adolescence, lower limb dystonia initially
- Diurnal variation — symptoms worse in evening, better in morning after sleep (pathognomonic)
- May have parkinsonism features
- Levodopa trial: dramatic and sustained response at low doses
- A levodopa trial should be performed in every young-onset generalised dystonia patient before any other diagnosis is accepted
4. Neurodegeneration with Brain Iron Accumulation (NBIA)
| Type | Gene | Hallmark |
|---|---|---|
| PKAN (NBIA-1) | PANK2 | "Eye of the tiger" sign on T2 MRI in globus pallidus |
| MPAN | C19orf12 | |
| BPAN | WDR45 | X-linked; females; regression then parkinsonism |
| FA2H | Fatty acid hydroxylase |
PKAN: onset in childhood/adolescence, generalised dystonia + rigidity, oromandibular dystonia, retinal degeneration (in classic form), acanthocytosis.
5. Biotin-Responsive Basal Ganglia Disease (BBGD)
- SLC19A3 mutation; AR
- Episodic encephalopathy triggered by febrile illness → dystonia, dysarthria, dysphagia, ophthalmoplegia
- MRI: bilateral caudate + putaminal lesions
- Responds to biotin + thiamine — treatable
Investigations — Stepwise Approach
Step 1: Mandatory Screening (Never Skip)
| Test | Target |
|---|---|
| MRI brain (T1, T2, FLAIR, SWI) | Structural/degenerative, iron (PKAN), Wilson changes |
| Serum ceruloplasmin | Wilson disease |
| 24-hr urinary copper | Wilson disease |
| Slit-lamp examination | Kayser-Fleischer rings (Wilson) |
| Liver function tests | Wilson (hepatic involvement) |
| Levodopa trial | DRD — must be tried in ALL young-onset dystonia |
Step 2: If Step 1 Negative
| Test | Target |
|---|---|
| Serum copper | Wilson |
| Blood film (acanthocytes) | Neuroacanthocytosis, HARP syndrome |
| Lysosomal enzyme panel | Gangliosidoses, MLD |
| Very long chain fatty acids | Adrenoleukodystrophy |
| Urine amino acids + organic acids | Organic acidaemias (glutaric, methylmalonic) |
| Arylsulfatase A | MLD |
| Filipin staining / NPC1 gene | Niemann-Pick C |
| HPRT enzyme | Lesch-Nyhan |
| Uric acid | Lesch-Nyhan |
| Lactate, pyruvate | Mitochondrial disease |
Step 3: Genetic Testing
| Gene Panel | When |
|---|---|
| DYT1 (TOR1A) deletion | Young-onset generalised, no secondary cause found |
| DYT6 (THAP1) | Adolescent onset, prominent cervical/laryngeal involvement |
| GCH1 (DRD) | If levodopa trial response uncertain |
| PANK2 | "Eye of tiger" on MRI |
| ATP7B | Wilson disease confirmation |
| Whole exome sequencing | If targeted panel negative |
Treatment
Step 1: Levodopa Trial — ALWAYS FIRST
- Start 50–100 mg levodopa/day, titrate
- DRD: dramatic sustained response at low doses — diagnostic and therapeutic
- Even DYT1 patients may have partial response
Step 2: Oral Medications
| Drug | Mechanism | Use |
|---|---|---|
| Trihexyphenidyl (anticholinergic) | Blocks muscarinic receptors | High-dose (up to 20–30 mg/day in young) — most effective for primary generalised dystonia |
| Baclofen | GABA-B agonist | Adjunct; intrathecal baclofen for severe cases |
| Clonazepam | GABAergic | Adjunct |
| Tetrabenazine | Dopamine depleter | For hyperkinetic component |
| Valproate | GABAergic | Adjunct |
| D-penicillamine / Trientine / Zinc | Copper chelation | Wilson disease — specific treatment |
| Biotin + Thiamine | Cofactor replacement | BBGD |
Step 3: Deep Brain Stimulation (DBS)
- Globus pallidus interna (GPi) stimulation — treatment of choice for medically refractory primary generalised dystonia
- Most effective in DYT1 (TOR1A mutation) — 50–90% improvement in BFMDRS
- Less effective in secondary dystonias
- Consider after adequate trial of medical therapy
Most Important Teaching Points
Sources: Localization in Clinical Neurology 8e; Adams & Victor's Principles of Neurology 12e; Harrison's Principles of Internal Medicine 22E
Amoebic meningoencephalitis
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Comprehensive data collected. Here is the full structured review:
Amoebic Meningoencephalitis
Overview
Four genera of free-living environmental amoebae — not requiring a human host — can cause CNS infection:
| Genus | CNS Syndrome |
|---|---|
| Naegleria fowleri | Primary Amoebic Meningoencephalitis (PAM) |
| Acanthamoeba spp. | Granulomatous Amoebic Encephalitis (GAE) + Keratitis |
| Balamuthia mandrillaris | Granulomatous Amoebic Encephalitis (GAE) |
| Sappinia pedata | GAE (single case reported) |
1. PRIMARY AMOEBIC MENINGOENCEPHALITIS (PAM)
Caused by Naegleria fowleri
Organism
- Thermophilic free-living amoeba; thrives in warm freshwater (lakes, rivers, poorly maintained pools, neti pots, waterslides)
- Exists in three forms: trophozoite (amoeboid) → flagellate (transient, non-feeding) → cyst
- Only trophozoite is pathogenic
Epidemiology
- Worldwide; ~381 cases globally through 2022; 0–8 cases/year in USA
- 75% male; median age 14 years (range 1 month–85 years)
- 91% had recreational freshwater exposure
- Cases rising in northern US states (climate change)
- Prevalence ~2.6 cases per million exposures
Pathogenesis
Warm freshwater inhaled nasally
↓
Trophozoites enter olfactory neuroepithelium
↓
Migrate along olfactory nerve → cribriform plate
↓
Enter CNS, bypass blood-brain barrier
↓
Fulminant necrotizing hemorrhagic encephalitis
↓
Neutrophilic response — olfactory bulbs/frontal lobes first
↓
Diffuse cerebral edema, uncal herniation → death (median <10 days)
Clinical Features
| Feature | Details |
|---|---|
| Incubation | 2–15 days |
| Fever | High, universal |
| Headache | Severe |
| Nausea/vomiting | Present early |
| Meningismus | Usually present (mimics bacterial meningitis) |
| Photophobia | Common |
| CN III, IV, VI palsies | Frequent |
| Seizures | Progressive |
| Coma | Rapid progression |
| Uncal herniation | Terminal event |
| Death | ~10 days from exposure; mortality >97% |
CSF Findings
| Parameter | Result |
|---|---|
| Opening pressure | Elevated |
| Appearance | Haemorrhagic / purulent |
| WBC | High — neutrophilic (up to 20,000/µL) |
| Protein | Elevated |
| Glucose | Low |
| Gram stain / culture | Negative for bacteria |
| Wet mount | Motile trophozoites — diagnostic |
Critical: CSF must be examined immediately as a fresh wet preparation — trophozoites are motile and die if refrigerated. This is the bedside diagnostic.
Diagnosis
| Test | Notes |
|---|---|
| Fresh CSF wet mount | Motile trophozoites (10–35 µm) — diagnostic |
| CDC PCR | Most sensitive; call CDC (770-488-7100) |
| Histochemical staining of biopsy | Available via CDC |
| MRI | Obliteration of cisterns, diffuse enhancement; olfactory bulb involvement |
| Serology | Not useful — antibodies found in healthy adults |
Treatment (Medical Emergency)
All 7 worldwide survivors received this combination:
| Drug | Role |
|---|---|
| Miltefosine (Impavido) | Central — all survivors received this; alkylphosphocholine; crosses BBB |
| Liposomal Amphotericin B (IV + intrathecal) | Primary amoebicidal |
| Fluconazole | Synergistic |
| Azithromycin | Synergistic |
| Rifampin | Synergistic |
| Dexamethasone | Neuroinflammation / cerebral edema |
| Therapeutic hypothermia | Associated with survival |
Contact CDC Emergency Operations: (770) 488-7100 — for miltefosine access and PCR diagnosis
2. GRANULOMATOUS AMOEBIC ENCEPHALITIS (GAE)
Caused by Acanthamoeba spp. and Balamuthia mandrillaris
These two share the GAE syndrome but differ in key ways:
Comparison: Acanthamoeba vs Balamuthia
| Feature | Acanthamoeba | Balamuthia mandrillaris |
|---|---|---|
| Host | Usually immunocompromised | Can affect immunocompetent |
| Risk factors | AIDS, transplant, steroids, lymphoma, lupus | Young age, Hispanic ethnicity, soil/water exposure |
| Entry route | Inhalation or skin contact → haematogenous | Percutaneous or mucous membrane → haematogenous |
| Onset | Subacute/chronic (weeks to months) | Subacute — weeks to months |
| Skin lesions | Cutaneous ulcers/nodules (especially AIDS) | Face/trunk/extremity skin lesions — early clue |
| CNS | Mimics space-occupying lesion | Focal deficits, seizures, headache, fever |
| CSF | Mononuclear pleocytosis; elevated protein | Mononuclear or neutrophilic pleocytosis; elevated protein; normal-low glucose |
| Amoebae in CSF | Rare | Rarely isolated |
| Brain imaging | Multiple hypodense/enhancing lesions; mimics toxoplasmosis | Multiple hypodense lesions |
| Diagnosis | Biopsy — trophozoites + cysts; culture on E. coli-seeded agar | CSF, biopsy; CDC PCR; fluorescent antibody |
| Transplant transmission | Less documented | Yes — 3 clusters 2009–2012 |
| Survival | Very rare; accelerated course in AIDS/transplant (3–40 days) | 9 survivors in USA with aggressive treatment |
| Treatment | Miltefosine + combination | Pentamidine + flucytosine + sulfadiazine + macrolides + miltefosine |
Acanthamoeba Keratitis (Separate Syndrome)
- Contact lens wearers (major risk — especially with homemade saline, swimming in lenses)
- Annular paracentral corneal ring — characteristic sign
- Irregular polygonal double-walled cysts on corneal scraping (see image below)
- Treatment: polyhexamethylene biguanide (0.2%) + propamidine isethionate (0.1%) drops; severe cases need keratoplasty
- Cysts are drug-resistant — difficult to treat
Side-by-Side Comparison: PAM vs GAE
| Feature | PAM (Naegleria) | GAE (Acanthamoeba / Balamuthia) |
|---|---|---|
| Onset | Acute/fulminant | Subacute/chronic |
| Host | Healthy, young, male | Immunocompromised (Acanthamoeba); any (Balamuthia) |
| Exposure | Warm freshwater swimming/nasal | Skin/soil/air → haematogenous |
| Entry | Olfactory neuroepithelium | Haematogenous from skin/lung/sinus |
| Pathology | Necrotizing haemorrhagic encephalitis | Granulomatous encephalitis |
| CSF | Neutrophilic, haemorrhagic, motile trophozoites | Mononuclear; organisms rarely seen |
| Meningismus | Present (like bacterial meningitis) | Variable; less prominent |
| MRI | Diffuse oedema, cisternal obliteration | Multiple focal lesions (mimic tumour/abscess/toxoplasmosis) |
| Diagnosis | Fresh CSF wet mount (motile trophozoites) | Brain biopsy (trophozoites + cysts) |
| Course | Death in <10 days | Weeks to months |
| Mortality | >97% | >95% |
| Key treatment | Miltefosine + AmB + FLU + AZI + RIF + Dexa | Miltefosine + organism-specific combinations |
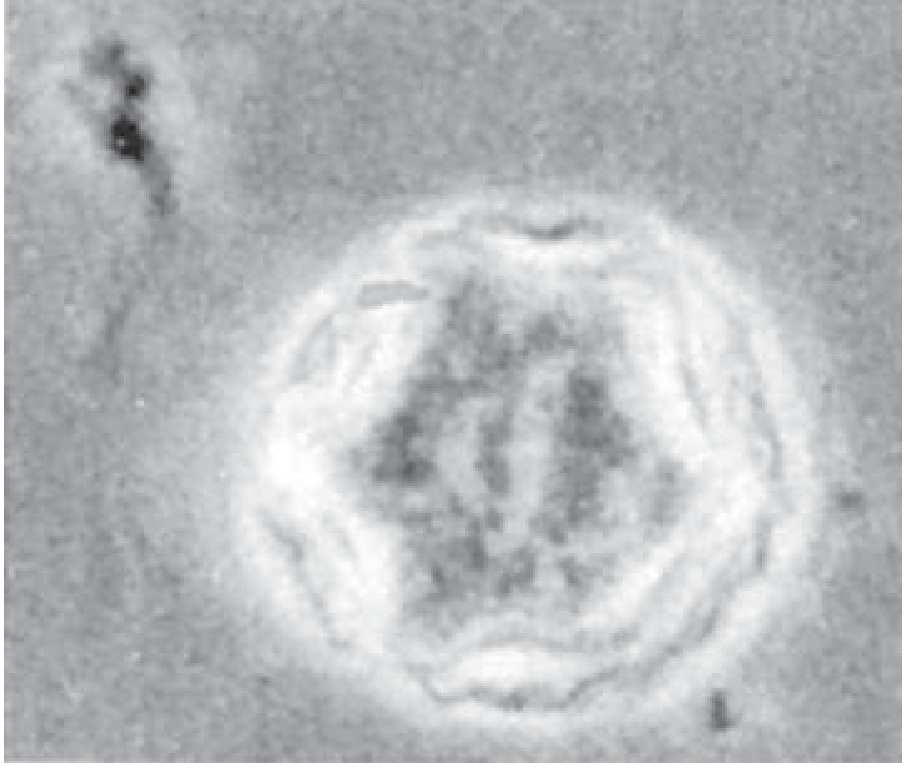
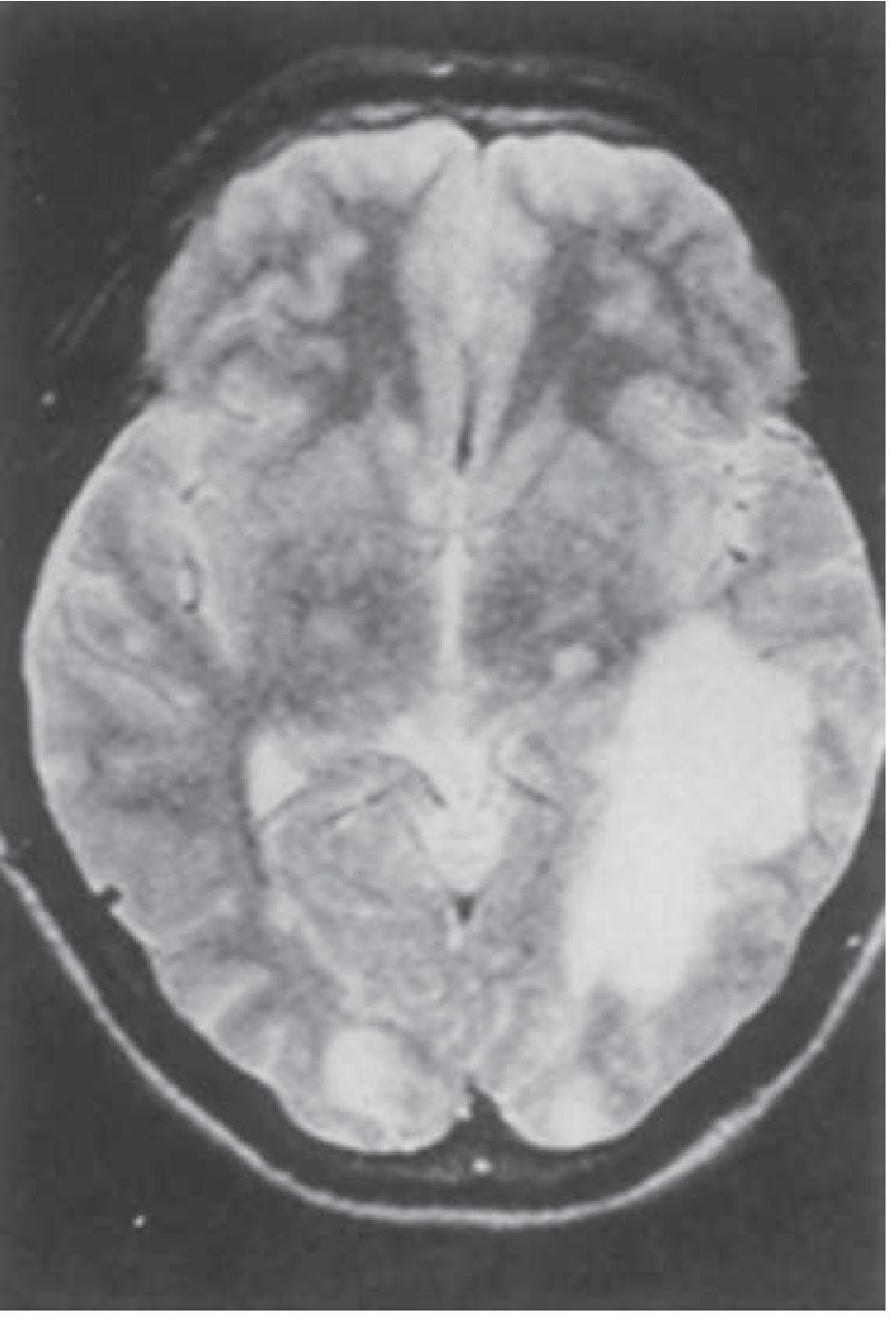
Pathological Images
Acanthamoeba castellani double-walled cyst (phase-contrast microscopy) — characteristic irregular polygonal shape:
Balamuthia mandrillaris GAE — MRI showing large parieto-occipital lesion and smaller satellite lesions:
When to Suspect Each
Young healthy person + warm freshwater swimming + acute meningitis-like illness
→ PAM (Naegleria) — CSF wet mount STAT
Immunocompromised + subacute focal brain lesions + skin nodules/ulcers
→ GAE (Acanthamoeba) — brain biopsy
Immunocompetent or immunocompromised + skin lesion on face + subacute encephalopathy
+ soil/water exposure or organ transplant recipient
→ GAE (Balamuthia) — CDC PCR + biopsy
Contact lens wearer + painful red eye + annular corneal ring
→ Acanthamoeba keratitis — corneal scraping for cysts
Key Points for Exams
- PAM = olfactory route (Naegleria); GAE = haematogenous route (Acanthamoeba, Balamuthia)
- PAM mimics bacterial meningitis; GAE mimics brain tumour or toxoplasmosis
- Diagnosis of PAM: motile trophozoites on fresh CSF wet mount (never refrigerate)
- Miltefosine is the most important drug for all three — included in all 7 PAM survivors
- Serology is not useful for PAM (antibodies found in healthy adults)
- Acanthamoeba keratitis: contact lens wearers; cysts resist treatment — high recurrence
- Balamuthia is the only free-living amoeba that regularly causes GAE in immunocompetent individuals
Sources: Harrison's Principles of Internal Medicine 22E; Robbins & Kumar Basic Pathology; Kaplan & Sadock's Comprehensive Textbook of Psychiatry; Medical Microbiology 9e; Tintinalli's Emergency Medicine
Hypertrophic olivary degeneration
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Searching PubMed
"hypertrophic olivary degeneration" MRI clinical
Finding Sources
Finding Sources
Reading File
Reading Article
PMID: 31026459
Searching Images
hypertrophic olivary degeneration MRI inferior olive T2
Finding Sources
Loading Image
Loading Image
Excellent images obtained. Here is the comprehensive review:
Hypertrophic Olivary Degeneration (HOD)
Definition
Hypertrophic Olivary Degeneration (HOD) is a rare form of trans-synaptic (transaxonal) degeneration of the inferior olivary nucleus (ION) in the medulla oblongata, occurring secondary to lesions disrupting the Guillain-Mollaret triangle (GMT).
Unlike most degenerative processes where neurons atrophy and shrink, the ION paradoxically enlarges — hence "hypertrophic" rather than atrophic degeneration. This is because the dominant pathological process is astrocytic hypertrophy and vacuolation of olivary neurons, not simple neuronal loss.
First described by Oppenheim in post-mortem studies; the enlarged ION was first correlated with clinical palatal myoclonus by Guillain and Mollaret.
The Guillain-Mollaret Triangle (GMT) — The Anatomical Key
HOD results from disruption of this three-cornered circuit:
MIDBRAIN
Red Nucleus (ipsilateral)
/ \
Central Tegmental Brachium Conjunctivum
Tract (CTT) (Superior Cerebellar Peduncle)
↓ ↑
MEDULLA CEREBELLUM
Inferior Olivary Dentate Nucleus
Nucleus (ION) (contralateral)
(ipsilateral)
↓ __________________|
Inferior Cerebellar
Peduncle (climbing fibers)
Three limbs of the triangle:
| Limb | Pathway | Direction |
|---|---|---|
| 1. Central Tegmental Tract (CTT) | Red nucleus → Inferior olive | Descending (ipsilateral) |
| 2. Superior Cerebellar Peduncle | Dentate nucleus → Red nucleus | Ascending (crosses at midbrain) |
| 3. Inferior Cerebellar Peduncle | Inferior olive → Dentate nucleus (climbing fibers) | Ascending (crosses) |
Lesion anywhere in the GMT → ION disinhibition → olivary pseudohypertrophy
Pathophysiology
Why Does the Olive Enlarge (Not Atrophy)?
When the CTT or the dentato-rubro pathway is disrupted:
- Deafferentation of the ION from its inhibitory inputs
- ION neurons undergo vacuolar change and astrocytic hypertrophy (not neuronal loss initially)
- Gap junctions between olivary neurons become upregulated → rhythmic synchronized discharges
- These discharges propagate via climbing fibers → cerebellar cortex → palatal muscles → palatal tremor
- Over months to years → eventual neuronal loss and olivary atrophy (late stage)
Timeline of MRI Changes
| Time from lesion | MRI finding |
|---|---|
| 0–1 month | No olivary change (normal MRI) |
| 1–6 months | T2 hyperintensity appears in ION (earliest sign) |
| 6 months–3–4 years | T2 hyperintensity + enlargement of ION (classic HOD) |
| >3–4 years | Enlargement resolves; T2 hyperintensity may persist or also regress |
The latency of months between the causative lesion and onset of palatal tremor / MRI changes is a hallmark of HOD.
Causes of HOD — Lesions Disrupting the GMT
Most Common
| Cause | Notes |
|---|---|
| Brainstem infarction (most common) | CTT in pons/midbrain; posterior circulation strokes |
| Brainstem haemorrhage | Pontine haemorrhage is the most common single cause |
| Cerebellar infarction/haemorrhage | Dentate nucleus involvement |
| Neurosurgical procedures | Posterior fossa surgery; tumour resection |
| Tumours | Brainstem glioma, metastases, cavernomas |
| Demyelination | MS plaques in CTT |
| Trauma | Diffuse axonal injury |
Less Common
- Cavernous malformation (haemosiderin deposition in GMT)
- Arteriovenous malformation
- Radiation injury
- Metabolic/toxic (e.g., Wernicke encephalopathy affecting midbrain)
Unilateral vs Bilateral HOD
This depends on which limb of the GMT is affected:
| Lesion Site | HOD Laterality | Explanation |
|---|---|---|
| Unilateral CTT (pons/midbrain) | Contralateral ION hypertrophy | CTT descends ipsilaterally to the olive; olive projects contralaterally to the dentate; the "disinhibited" side is contralateral to the CTT lesion |
| Unilateral dentate nucleus | Ipsilateral ION hypertrophy | Dentato-rubro-olivary pathway |
| Both CTT + dentate nucleus (or crossing fibres) | Bilateral HOD | More complex lesions |
Rule: With unilateral palatal tremor, the contralateral olive enlarges. — Adams & Victor's Neurology 12e
Clinical Features
Cardinal Feature: Symptomatic Palatal Tremor
- Rhythmic, 1–3 Hz contractions of the levator veli palatini (CN X)
- Persists during sleep (unlike essential palatal tremor)
- No auditory click (unlike essential palatal tremor)
- Onset: typically months after the causative lesion
Other Oculomotor Features
- Pendular nystagmus (synchronised with palatal movements)
- Oculopalatal myoclonus:
- Unilateral lesion: jerky, nystagmoid, oblique/rotatory eye movements
- Bilateral lesion: vertical to-and-fro pendular nystagmus
Other Movements
- Rhythmic contractions of pharynx, facial muscles, diaphragm, vocal cords, neck/shoulders
- Dentatorubral tremor — limb tremor from dentate/rubral involvement
- Cerebellar ataxia (from the primary lesion)
Neurological Deficits from Primary Lesion
- Hemiparesis, dysarthria, dysphagia — depending on brainstem lesion location
MRI Findings — The Diagnostic Signature
T2/FLAIR: Hyperintensity + enlargement of the inferior olivary nucleus in the medulla
T1: Hypointense (no enhancement with gadolinium — distinguishes from tumour)
No gadolinium enhancement — important to differentiate from:
- Brainstem neoplasm
- Demyelinating plaque
- Inflammatory lesion
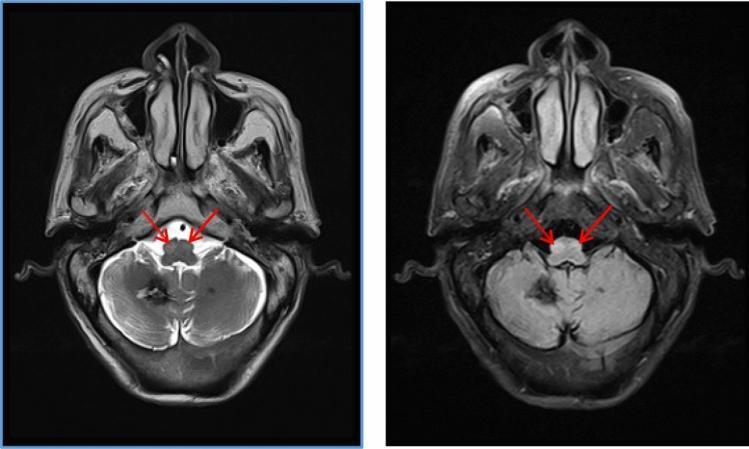
Classic MRI Appearance:
HOD on T2 MRI — bilateral inferior olive hyperintensity and enlargement (red arrows):
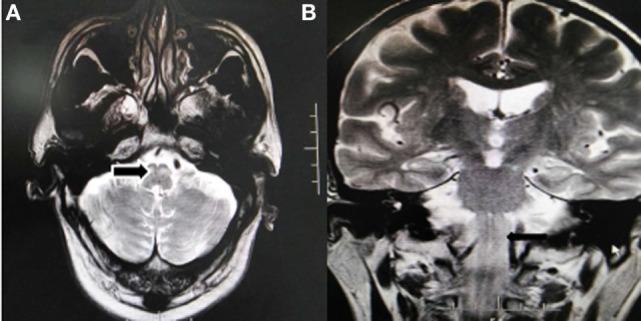
HOD axial and coronal T2 — unilateral enlargement of inferior olive (black arrows):
Differential Diagnosis of Inferior Olive T2 Signal
| Condition | Key Differentiator |
|---|---|
| HOD | No enhancement; enlarged; delayed onset after GMT lesion; palatal tremor |
| Brainstem tumour | Enhancement; mass effect; no latency |
| Demyelinating plaque (MS) | Other white matter lesions; may enhance acutely |
| Wernicke encephalopathy | Mammillary bodies + periaqueductal gray + thalamus also involved |
| Osmotic demyelination | Pons centrally; history of hyponatraemia correction |
| Brainstem infarction (acute) | DWI restriction; acute onset |
HOD vs Essential Palatal Tremor — Quick Recap
| Symptomatic Palatal Tremor (= HOD) | Essential Palatal Tremor | |
|---|---|---|
| Muscle | Levator veli palatini | Tensor veli palatini |
| Ear click | ❌ No | ✅ Yes |
| Sleep | Persists | Disappears |
| MRI olive | Enlarged + T2 bright | Normal |
| GMT lesion | ✅ Present | ❌ None |
| Onset after lesion | Months | No prior lesion |
Treatment
| Intervention | Notes |
|---|---|
| Gabapentin | Most effective (dramatic in some) |
| Clonazepam | 0.25–6 mg/day |
| Sodium valproate | 250–1000 mg/day |
| Memantine | Reported benefit (NMDA receptor modulation of olivary rhythmicity) |
| Botulinum toxin | Palatal injection — modest relief |
| Treat primary lesion | If vascular, manage risk factors |
| No cure | HOD itself is irreversible once established |
Summary
HOD = trans-synaptic degeneration of the inferior olive, occurring months after a lesion interrupts the Guillain-Mollaret triangle (red nucleus → central tegmental tract → inferior olive → dentate nucleus → back)MRI: T2/FLAIR hyperintensity + enlargement of ION, no gadolinium enhancement, delayed onset 1–6 months after causative lesionClinically: Symptomatic palatal tremor + pendular nystagmus ± rhythmic myoclonus of face/pharynx/diaphragmKey concept: The olive hypertrophies (not atrophies) because the process is astrocytic vacuolation and gap junction upregulation — not simple neuronal loss
Sources: Adams & Victor's Principles of Neurology 12e; Localization in Clinical Neurology 8e; Wang et al. Brain Research 2019 [PMID 31026459]
How to ask for history in dementia for all lobes
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
I now have sufficient data from Harrison's and Localization in Clinical Neurology to give a comprehensive, clinically structured answer. Here it is:
Taking History in Dementia — Lobe by Lobe
Principle
Each cerebral lobe subserves specific cognitive functions. When taking a history in dementia, you systematically ask questions that probe each lobe's function — mapping the pattern of deficits to the anatomical distribution of disease. This tells you not just that the patient has dementia, but what type, and guides investigation and management.
Always obtain history from both patient AND a reliable informant (caregiver/family) — the patient may lack insight (anosognosia), especially in frontal or parietal disease.
Framework: What Each Lobe Does and What to Ask
🔵 FRONTAL LOBE
Functions: Executive function, working memory, planning, sequencing, abstract reasoning, impulse control, personality, social behaviour, motor speech (Broca's area — dominant), motor cortex
Questions to Ask:
Executive / Planning:
- "Has he/she become disorganised? Does he/she struggle to plan a meal, manage money, or pay bills?"
- "Can he/she do tasks in the right order — like making tea (fill kettle, boil, pour)?"
- "Does he/she start tasks but not finish them?"
- "Can he/she switch between tasks, or does he/she get stuck on one thing?"
Behaviour / Personality Change:
- "Has his/her personality changed? Is he/she a different person from before?"
- "Has he/she become disinhibited — saying rude or inappropriate things, making sexual remarks, or behaving badly in public?"
- "Is he/she impulsive — spending money recklessly, grabbing food off others' plates?"
- "Has he/she lost social awareness — does he/she embarrass the family?"
Apathy:
- "Has he/she become very passive, lost initiative, stopped doing things he/she used to enjoy?"
- "Does he/she sit all day without doing anything unless prompted?"
- "Is there poverty of speech — speaking very little spontaneously?"
Compulsive / Stereotyped Behaviour:
- "Does he/she repeat the same actions or phrases over and over?"
- "Does he/she have rigid routines — insisting on eating the same food or following the same route?"
Hyperorality:
- "Does he/she put objects in the mouth? Eat excessively or indiscriminately?"
Motor Speech (Broca's / Non-fluent Aphasia):
- "Has his/her speech become halting, effortful, or telegraphic (short sentences)?"
- "Does he/she struggle to get words out even though he/she seems to know what to say?"
Suggests: Frontotemporal dementia (bvFTD), non-fluent/agrammatic PPA, PSP, CBD
🟠 TEMPORAL LOBE
Functions: Episodic memory (medial temporal — hippocampus), semantic memory (anterior temporal), language comprehension (Wernicke's — dominant), face and object recognition, auditory processing, emotion (amygdala)
Questions to Ask:
Episodic Memory (Hippocampus / Medial Temporal):
- "Does he/she repeat the same questions or stories within minutes — not realising they've already said it?"
- "Does he/she forget recent events — what happened yesterday, what he/she had for breakfast?"
- "Does he/she remember remote events (childhood, old friends) better than recent ones?"
- "Does he/she forget appointments, miss medications, forget names of recent acquaintances?"
- "Does he/she get lost in familiar places — e.g., going to the local shop?"
Semantic Memory (Anterior Temporal):
- "Has he/she lost knowledge of objects — not recognising what a 'kettle' or 'umbrella' is?"
- "Has he/she lost word meanings — uses words incorrectly or says 'that thing' instead of the name?"
- "Does he/she fail to recognise faces of famous people (e.g., politicians)?"
Language Comprehension (Wernicke's — dominant temporal):
- "Does he/she have difficulty understanding what is said to him/her?"
- "Does he/she give nonsensical answers to questions — as if talking about something different?"
- "Is his/her speech fluent but empty — lots of words but no meaning?"
Prosopagnosia (Face Recognition — non-dominant):
- "Does he/she fail to recognise familiar faces — even close family members?"
Auditory/Musical:
- "Any change in appreciation of music or sounds?"
Suggests: Alzheimer's disease (medial temporal first), LATE, semantic variant PPA (anterior temporal), Wernicke's aphasia in PPA
🟡 PARIETAL LOBE
Functions: Visuospatial processing (non-dominant), attention (bilateral), calculation, writing, reading (dominant), praxis (dominant — left), sensory integration, body schema
Questions to Ask:
Visuospatial (Non-dominant — Right Parietal):
- "Does he/she get lost in familiar places — his/her own house, or the route to the local shop or temple?"
- "Does he/she have difficulty parking the car, judging distances, or navigating in a mall?"
- "Does he/she bump into furniture or objects on one side?"
- "Does he/she have difficulty recognising objects or faces even with normal vision?"
Apraxia (Dominant — Left Parietal):
- "Has he/she lost the ability to use everyday tools correctly — using a toothbrush, a key, a fork, a lighter?"
- "Has he/she become clumsy with buttons, zips, or shoelaces?"
- "Does he/she have difficulty imitating gestures (e.g., waving goodbye)?"
Acalculia / Agraphia / Alexia (Dominant):
- "Can he/she still manage money — count change, do simple arithmetic?"
- "Has his/her writing become abnormal?"
- "Can he/she still read — newspaper, messages?"
Gerstmann Syndrome (Dominant Angular Gyrus: agraphia + acalculia + finger agnosia + left-right disorientation):
- "Does he/she confuse left and right?"
- "Can he/she identify individual fingers?"
Neglect (Non-dominant Parietal):
- "Does he/she ignore one side of the body or environment — only eating food from one side of the plate? Only shaving/grooming one side of the face?"
Dressing Apraxia (Non-dominant):
- "Does he/she have difficulty dressing — putting clothes on the wrong way, inability to work out how garments go on?"
Suggests: Posterior cortical atrophy (PCA — Alzheimer's variant), corticobasal degeneration, DLB (posterior parietal)
🟢 OCCIPITAL LOBE
Functions: Primary visual processing, visual recognition, colour, motion perception
Questions to Ask:
Visual Hallucinations:
- "Does he/she see things that are not there — people, animals, patterns?"
- "How vivid are they? Are they frightening or not?"
- "Do they occur in low light conditions?"
Visual Object Agnosia:
- "Can he/she recognise objects just by looking at them (with normal vision)?"
- "Does he/she mistake objects for other things?"
Colour / Motion:
- "Any difficulty recognising colours or perceiving moving objects?"
Cortical Blindness / Anton Syndrome:
- "Is he/she unaware of being blind — denying the visual problem even when it is obvious?"
Suggests: DLB (vivid visual hallucinations), posterior cortical atrophy, CJD, occipital strokes
⚫ LIMBIC / CINGULATE
Functions: Emotion, motivation, mood, social cognition
Questions to Ask:
- "Has he/she become emotionally flat — no joy, no sadness?"
- "Is he/she socially withdrawn, no longer interested in family or friends?"
- "Is he/she irritable, anxious, or depressed — a new change from baseline?"
⚪ SUBCORTICAL / WHITE MATTER
Functions: Processing speed, attention, working memory, motor coordination
Questions to Ask:
- "Has he/she slowed down mentally — takes much longer to answer questions or complete tasks?"
- "Is there forgetfulness that improves with prompting (subcortical — unlike cortical where cueing doesn't help)?"
- "Has there been change in gait — shuffling, falls, urinary incontinence (NPH triad)?"
Suggests: Vascular dementia, Binswanger's, NPH, Huntington's, PSP
Putting It Together: Pattern Recognition
| Predominant Lobe | Earliest Symptoms | Common Diagnosis |
|---|---|---|
| Medial temporal | Episodic memory loss, repetition | Alzheimer's disease, LATE |
| Frontal | Personality, behaviour, disinhibition, apathy | FTD (bvFTD), PSP |
| Dominant temporal (anterior) | Word meaning loss, fluent empty speech | Semantic PPA |
| Dominant temporal (posterior) | Comprehension failure, fluent paraphasia | Wernicke's/logopenic PPA |
| Dominant frontal | Effortful, halting speech | Non-fluent PPA, CBS |
| Parietal + occipital | Visuospatial, apraxia, visual hallucinations | PCA, DLB, CBS |
| Subcortical + frontal | Slowing, gait, incontinence | Vascular, NPH |
| Multifocal/rapid | Any combination + rapid progression | CJD, autoimmune encephalitis |
Additional Screening History (Always Ask)
| Domain | Questions |
|---|---|
| Onset | Sudden (vascular) vs gradual (degenerative) vs fluctuating (DLB) |
| Course | Progressive / stepwise (vascular) / relapsing |
| ADLs | Dressing, bathing, cooking, driving, finances, medications |
| Sleep | REM sleep behaviour disorder (punching/kicking in sleep) → DLB/PD |
| Falls | Repeated early falls → PSP |
| Hallucinations | Visual hallucinations early → DLB |
| Drugs | Anticholinergics, benzodiazepines, opioids |
| Risk factors | Hypertension, diabetes, smoking (vascular), family history |
| Mood | Depression can mimic and worsen dementia |
| Occupational history | Toxic exposures; head trauma (CTE) |
Memory Aid: "FLPOT" — 5 Lobes × Signature Symptoms
| Lobe | Signature Question |
|---|---|
| Frontal | "Has his personality changed? Does he do inappropriate things?" |
| Limbic/Temporal | "Does he repeat the same question within minutes?" |
| Parietal | "Does he get lost in familiar places? Can he still dress himself?" |
| Occipital | "Does he see things that aren't there?" |
| Temporal (semantic) | "Has he lost the meaning of words? Does he recognise familiar faces?" |
Sources: Harrison's Principles of Internal Medicine 22E (Table 33-4, Functional Anatomy of Dementias); Localization in Clinical Neurology 8e; Adams & Victor's Principles of Neurology 12e
Dystonia
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.