Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathophysiology mechanism diagram
Reading File
Loading Image
I now have comprehensive information from multiple authoritative sources. Let me compile the full answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
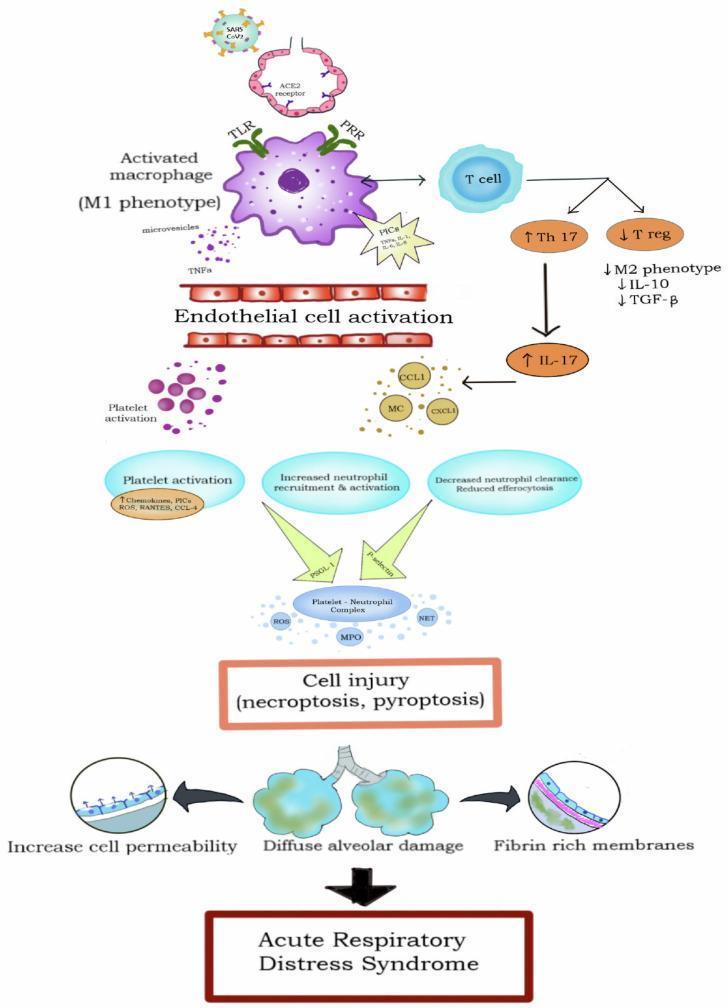
1. Definition and Triggers
ARDS is a syndrome of non-cardiogenic pulmonary edema caused by diffuse inflammatory injury to the alveolar-capillary barrier. The Berlin definition requires:
- Bilateral infiltrates on CXR/CT not fully explained by effusions or collapse
- PaO₂/FiO₂ (P/F) ratio < 300 mmHg on PEEP ≥ 5 cmH₂O (mild <300, moderate <200, severe <100)
- Onset within 7 days of a known clinical insult
- Not fully explained by cardiac failure or fluid overload
Common precipitants (direct/indirect lung injury):
| Direct (pulmonary) | Indirect (extrapulmonary) |
|---|---|
| Pneumonia, aspiration | Sepsis (most common overall) |
| Pulmonary contusion | Severe pancreatitis |
| Inhalation injury | Major trauma/hemorrhage |
| Near-drowning | Transfusion (TRALI) |
The initiating insult reaches the lung either via the airways (direct) or via the bloodstream (indirect). — Fishman's Pulmonary Diseases and Disorders, p. 2480
2. Core Pathophysiology: Breakdown of the Alveolar-Capillary Barrier
Step 1 — Initiating Injury and Innate Immune Activation
The inciting insult activates alveolar macrophages and circulating monocytes through pattern recognition receptors (TLRs, PRRs), shifting them to the M1 pro-inflammatory phenotype. They release a cascade of pro-inflammatory cytokines:
- TNF-α, IL-1β, IL-6, IL-8 (CXCL8) — amplify inflammation and recruit neutrophils
- IL-17 (via Th17 skewing) — further promotes neutrophil influx
- Anti-inflammatory regulators (IL-10, TGF-β, T-regulatory cells) are simultaneously suppressed
This inflammatory response begins before ARDS is clinically recognizable and peaks within 1–3 days. — Fishman's Pulmonary Diseases and Disorders, p. 2480
Step 2 — Neutrophil Sequestration (Central Effector)
Neutrophils are the principal mediators of alveolar injury. IL-8 and other chemokines attract massive neutrophil recruitment into the pulmonary vasculature and alveolar spaces. Activated neutrophils cause tissue damage through:
- Reactive oxygen species (ROS) — oxidative injury to the endothelial and epithelial membranes
- Proteases (elastase, matrix metalloproteinases) — degrade basement membrane and extracellular matrix
- Neutrophil extracellular traps (NETs) — fibrous chromatin scaffolds that amplify inflammation
- Myeloperoxidase (MPO) — generates hypochlorous acid, causing direct cellular necrosis
Activated neutrophils also degrade surfactant apoproteins via proteolysis and oxidant radical mechanisms. — Murray & Nadel's Textbook of Respiratory Medicine, p. 3115
Step 3 — Platelet-Neutrophil Complexes and Coagulation
Endothelial activation triggers platelet activation. Platelets bind neutrophils via P-selectin/PSGL-1 interactions, forming platelet-neutrophil complexes that amplify ROS and MPO release, intensifying cellular injury. Concurrent intravascular coagulation deposits fibrin within alveolar spaces, worsening surfactant inhibition and promoting hyaline membrane formation.
Step 4 — Endothelial and Epithelial Barrier Disruption
Cytokines, ROS, and proteases cooperate to disrupt both barriers:
- Vascular endothelium: cytoplasmic swelling, loss of tight junction integrity → increased vascular permeability → protein-rich fluid floods the interstitium
- Alveolar epithelium (the more critically damaged layer): widespread destruction of Type I pneumocytes (which cover ~95% of the alveolar surface) → denudement of basement membranes → loss of the primary defense against alveolar flooding
Because the protein osmotic gradient across the damaged barriers is abolished, even normal hydrostatic pressure drives fluid and protein into the alveolar space unopposed. In sepsis, high cardiac output may further increase microvascular hydrostatic pressure, worsening edema. — Fishman's Pulmonary Diseases and Disorders, p. 2479
3. Pathologic Phases
Phase 1 — Exudative (Days 1–7)
- Widespread alveolar and interstitial edema, hemorrhage, and capillary congestion
- Destruction of Type I pneumocytes; adjacent alveoli may appear normal ("geographic" pattern)
- Hyaline membranes form: composed of precipitated plasma proteins, fibrin, cellular debris, and necrotic epithelial remnants — this is the pathologic signature termed Diffuse Alveolar Damage (DAD)
- Polymorphonuclear leukocytes embedded in hyaline membranes
- The interstitium is widened by edema, leukocytes, platelets, fibrin, and debris (especially peribronchovascular cuffs)
Phase 2 — Proliferative (Days 7–21)
- Partial reabsorption of edema fluid
- Type II pneumocyte hyperplasia (cuboidal cells repopulate the alveolar surface as a repair response)
- Fibroblast infiltration and fibrin deposition; air-blood barrier thickened
- Pulmonary vascular disruption; reduced microvascular surface area
Phase 3 — Fibrotic (Weeks to Months, not universal)
- Progressive interstitial fibrosis in some patients
- Linked to prolonged mechanical ventilation and inability to resolve the inflammatory response
- Survivors with this phase have significantly impaired quality of life — Fishman's Pulmonary Diseases and Disorders, p. 2480
4. Surfactant Dysfunction
The injured lung releases substances that interfere with normal alveolar surface tension. Several converging mechanisms impair surfactant:
- Activated neutrophils degrade surfactant apoproteins (SP-B, SP-C) by proteolysis and oxidant-radical mechanisms
- Edema proteins (fibrin, fibrinogen, albumin) directly inhibit surfactant function
- Phospholipase A2 (released in pancreatitis and other states) enzymatically degrades surfactant phospholipids
- Intraalveolar coagulation compounds depletion
BAL fluid from ARDS patients is abnormal in both chemical composition and functional activity. The result is atelectasis, alveolar collapse, and dramatically increased work of breathing. — Murray & Nadel's Textbook of Respiratory Medicine, p. 3115
5. Physiologic Consequences
| Abnormality | Mechanism |
|---|---|
| Severe hypoxemia | Intrapulmonary shunting (flooded alveoli perfused but not ventilated) + V/Q mismatch |
| Reduced lung compliance | Loss of ventilated alveolar units; atelectasis; reduced FRC |
| Elevated dead space | Ventilated alveoli not perfused (microvascular disruption, thrombosis) |
| Hypercapnia/↑ minute ventilation | Dead space fraction elevated; minute ventilation rises to ~12–16 L/min |
| Increased airflow resistance | Reduced lung volumes; bronchospasm |
| Pulmonary hypertension | Hypoxic vasoconstriction + microvascular obliteration |
The "sponge" lung concept: the heavy, edematous ARDS lung is non-homogeneous — consolidation is gravity-dependent. This means a large portion of the lung is recruitable with PEEP, but prone to overdistension injury in already-aerated regions. — Goldman-Cecil Medicine, p. 1066
6. Ventilator-Induced Lung Injury (VILI) — A Secondary Mechanism
Mechanical ventilation itself can perpetuate injury:
- Volutrauma/barotrauma: overdistension of remaining aerated alveoli causes mechanical injury, releasing pro-inflammatory cytokines (biotrauma)
- Atelectrauma: repeated opening and closing of collapsed alveoli generates shear stress
- Both pathways release leukotrienes, prostaglandins, macrophage-derived ROS, and IL-8 into the systemic circulation, potentially causing distal organ dysfunction (multi-organ failure) — Goldman-Cecil Medicine, p. 1067
This provides the physiologic rationale for lung-protective ventilation (6 mL/kg predicted body weight tidal volumes, plateau pressure ≤ 30 cmH₂O).
Summary Cascade
Inciting insult (sepsis/pneumonia/aspiration/trauma)
↓
Alveolar macrophage activation → TNF-α, IL-1β, IL-6, IL-8
↓
Neutrophil recruitment to lung → ROS, proteases, NETs, MPO
↓
Platelet-neutrophil complexes → amplified oxidative injury
↓
Endothelial injury → ↑ vascular permeability → protein-rich edema
↓
Epithelial injury (Type I pneumocyte loss) → alveolar flooding
↓
Surfactant inactivation → atelectasis
↓
DAD: hyaline membranes, edema, hemorrhage, hyaline membranes
↓
Intrapulmonary shunt + ↓ compliance + ↑ dead space
↓
Refractory hypoxemia → ARDS
Key sources:
- Fishman's Pulmonary Diseases and Disorders, pp. 2479–2481
- Murray & Nadel's Textbook of Respiratory Medicine, pp. 3115–3116
- Goldman-Cecil Medicine, pp. 1066–1067
- Sabiston Textbook of Surgery, p. 940
This is a shared conversation. Sign in to Orris to start your own chat.