Acute kidney Injury
Reading File
Finding Sources
Searching PubMed
"acute kidney injury"[MeSH Terms] AND management
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Now I have comprehensive material to write a thorough, well-structured AKI review. Let me now compile the full answer.
Acute Kidney Injury (AKI)
Definition
Acute kidney injury is a clinical syndrome defined by a rapid decline in the kidney's ability to clear waste products, occurring within approximately 1 week of an injurious stimulus. The 2012 KDIGO (Kidney Disease: Improving Global Outcomes) criteria are the current gold standard:
AKI is defined by ANY ONE of the following:
- Rise in serum creatinine (sCr) by ≥0.3 mg/dL within 48 hours, OR
- Rise in sCr to ≥1.5 times baseline within 7 days, OR
- Urine output <0.5 mL/kg/hour for ≥6 hours
The term "Acute Kidney Disease" (AKD) was introduced by KDIGO to capture cases where creatinine rises evolve over periods longer than 7 days and thus do not meet the formal AKI definition. - NKF Primer on Kidney Diseases, 8e
KDIGO Staging
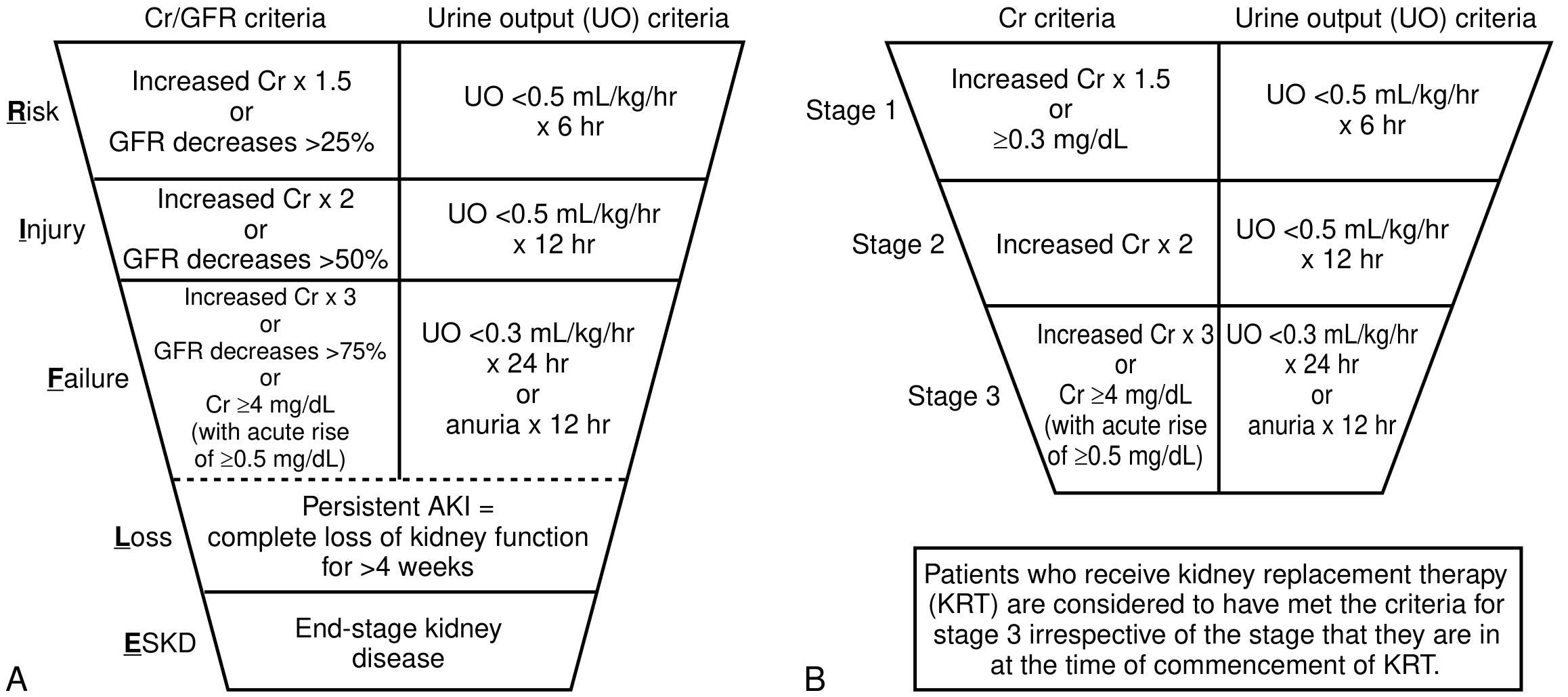
| Stage | Serum Creatinine | Urine Output |
|---|---|---|
| 1 | 1.5-1.9x baseline OR ≥0.3 mg/dL increase | <0.5 mL/kg/h for 6-12 hrs |
| 2 | 2.0-2.9x baseline | <0.5 mL/kg/h for ≥12 hrs |
| 3 | ≥3.0x baseline OR sCr ≥4.0 mg/dL OR initiation of KRT | <0.3 mL/kg/h for ≥24 hrs OR anuria ≥12 hrs |
Patients receiving kidney replacement therapy (KRT) are considered to have met Stage 3 criteria regardless of the creatinine level at the time of KRT initiation. - Goldman-Cecil Medicine, Table 106-1
The earlier RIFLE classification used a funnel-like staging system (Risk, Injury, Failure, Loss, ESKD) that preceded KDIGO but applied similar creatinine and urine output thresholds.
Epidemiology
-
~20-25% of hospitalized adults have an elevated serum creatinine
-
Up to 45% of ED admissions present with elevated sCr
-
55-65% of ICU patients develop AKI
-
Community-acquired AKI: annual incidence <1%
-
Prerenal azotemia accounts for 40% of hospital-acquired and 60-70% of community-acquired AKI cases
-
Intrinsic AKI is present in 5-6% of ED admissions and up to 60% of ICU patients
-
Intrinsic AKI requiring dialysis has increased substantially over the past three decades
-
Goldman-Cecil Medicine, p. 1241
Three Anatomic Categories
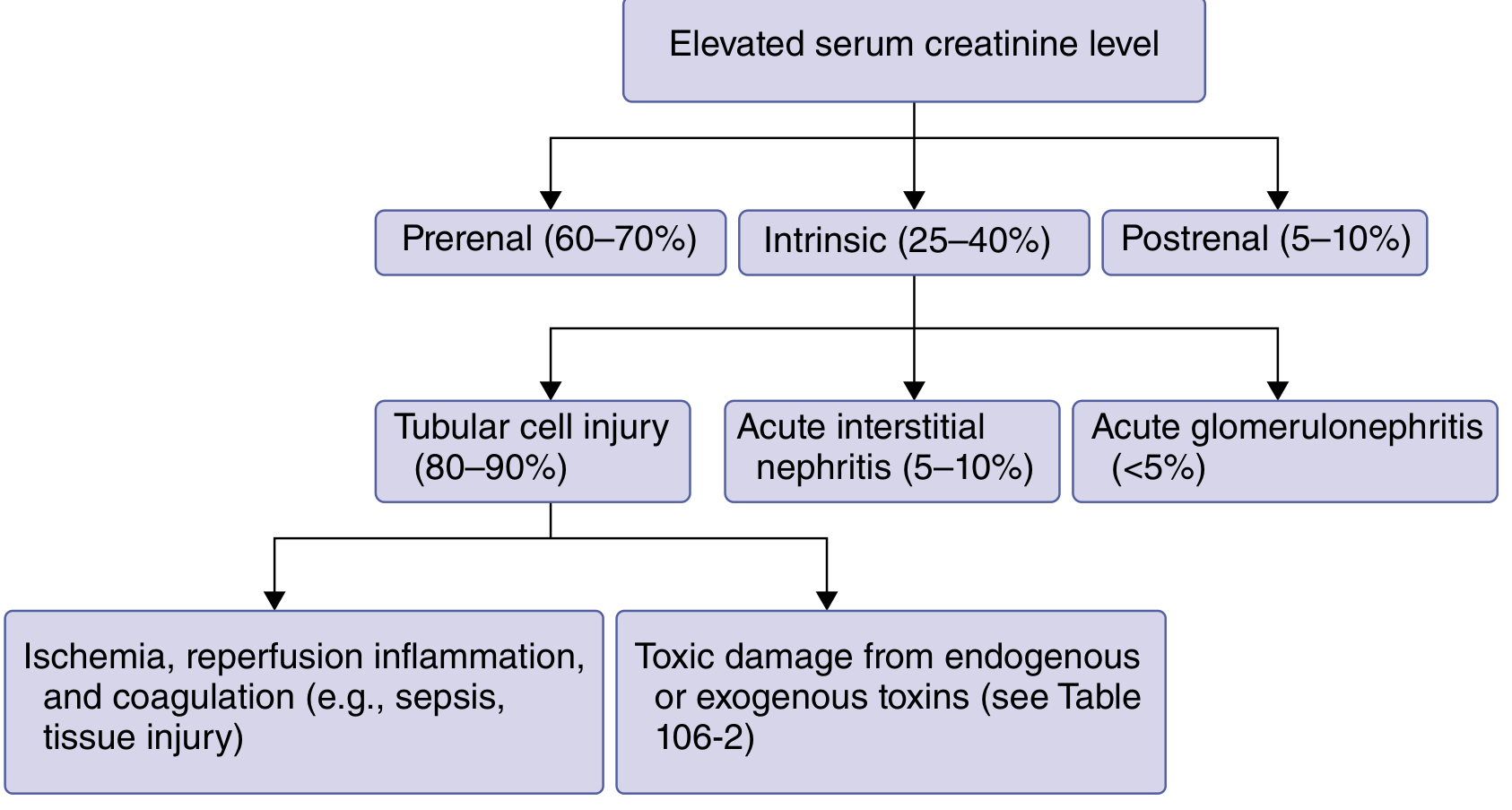
1. Prerenal AKI (60-70% of cases)
Mechanism: Decreased renal blood flow/perfusion without structural kidney damage.
Causes:
- True volume depletion: Limited oral intake, fever/sweating, burns, diarrhea, diuretics, hemorrhage
- Effective volume depletion: Heart failure, liver failure/cirrhosis, nephrotic syndrome
- Medications reducing GFR: NSAIDs, ACE inhibitors, angiotensin receptor blockers, cyclosporine, radiocontrast agents
Key features:
- BUN:Creatinine ratio >20:1 (increased urea reabsorption due to low effective arterial volume)
- FENa <1% in oliguric patients (intact tubular sodium reabsorption)
- Rapidly reversible with volume repletion within hours
- Urinary biomarkers of tubular injury not particularly elevated
FENa can be influenced by diuretics; in such cases, FEurea <35% is more reliable. - NKF Primer, p. 346
2. Intrinsic AKI (25-40% of cases)
An abrupt, sustained decline in function due to structural kidney damage - cannot be readily reversed for days to weeks.
Subtypes and frequencies:
- Tubular cell injury (80-90% of intrinsic AKI):
- Ischemic/ATN: Ischemia, reperfusion, inflammation, coagulation (e.g., sepsis, major surgery, cardiogenic shock)
- Toxic ATN: Endogenous toxins (myoglobin, hemoglobin, uric acid crystals, calcium oxalate) or exogenous (radiocontrast, aminoglycosides, vancomycin, amphotericin B, cisplatin, tacrolimus, cyclosporine)
- Acute Interstitial Nephritis (5-10%): Drug-induced (sulfonamides, penicillins, cephalosporins, NSAIDs, PPIs, checkpoint inhibitors), infections (Legionella, leptospirosis, SARS-CoV-2), autoimmune (SLE, Sjögren's)
- Acute Glomerulonephritis (<5%): Rapidly progressive GN, vasculitis
Molecular pathobiology: Activation of inflammatory, ischemic, and coagulation pathways; slow resolution due to vasoconstriction, tubular damage, cell death, and interstitial edema. Urinary biomarkers (NGAL, IL-18, KIM-1, TIMP-2, IGFBP7) are prominently elevated. - Goldman-Cecil Medicine, p. 1242
3. Postrenal AKI (5-10% of cases)
Mechanism: Obstruction of urine flow from kidneys to urethra.
Causes:
- Men: Benign prostatic hypertrophy (most common), bladder outlet obstruction
- Both sexes: Nephrolithiasis, pelvic malignancy, retroperitoneal fibrosis, neurogenic bladder
- Children: Congenital anatomic abnormalities
Relieving the obstruction can restore kidney function, but prolonged obstruction leads to tubular damage and may not be fully reversible.
Pathophysiology of AKI (ATN)
The proximal tubule and medullary thick ascending limb are most vulnerable due to high metabolic activity and limited oxygen supply (the medullary-cortical oxygen gradient). Following ischemia:
- Depletion of ATP → loss of cell polarity → detachment of tubular epithelial cells
- Cells form casts obstructing tubular flow → back-leak of filtrate
- Intense afferent arteriolar vasoconstriction (tubuloglomerular feedback via macula densa)
- Endothelial activation → inflammation, neutrophil and monocyte infiltration
- Reparative phase involves upregulation of genes for cell-cycle regulation, growth factors (KIM-1, NGAL), and cytokines
Diagnosis and Evaluation
Serum Creatinine - Limitations
sCr is widely available but has important limitations:
- Reflects GFR only at steady-state; there is a 24-72 hour lag before sCr stabilizes after an acute injury
- Influenced by age, sex, muscle mass, and catabolic state
- Volume resuscitation dilutes sCr, masking true AKI severity (the FACTT trial demonstrated fluid-corrected sCr identified more AKI cases)
- In the elderly and malnourished, lower creatinine generation means sCr rises more slowly even with severe injury
Urine Studies
| Test | Prerenal | ATN |
|---|---|---|
| FENa | <1% | >2% |
| FEUrea | <35% | >50% |
| Urine Na | <10 mEq/L | >40 mEq/L |
| Urine Osmolality | >500 mOsm/kg | <350 mOsm/kg |
| Urine sediment | Normal/hyaline casts | Muddy brown granular casts, tubular cells |
| BUN:Cr ratio | >20:1 | ~10-15:1 |
Urinary electrolytes should not be used in isolation - they are influenced by diuretics, sepsis, cirrhosis, and the phase of AKI when obtained. - NKF Primer, p. 347
Novel Biomarkers
| Biomarker | Source | Use |
|---|---|---|
| NGAL (Neutrophil Gelatinase-Associated Lipocalin) | Distal tubule | Early ATN detection (rises within hours) |
| KIM-1 (Kidney Injury Molecule-1) | Proximal tubule | Ischemic/toxic tubular injury |
| IL-18 | Proximal tubule | Predicts AKI progression and long-term mortality; AUC ~0.74-0.76 in cardiac surgery |
| TIMP-2 × IGFBP7 | Tubular cells | Cell-cycle arrest markers; FDA-cleared for AKI risk prediction |
| Cystatin C | All nucleated cells, freely filtered | Earlier GFR marker than creatinine |
| α1-Microglobulin | Proximal tubule reabsorption | AUC ~0.86 for predicting need for RRT |
Brenner and Rector's The Kidney, 2-Volume Set
Management
General Principles (Goldman-Cecil Medicine, p. 1243)
"Treatments involve removal of offending stimuli, correcting any obstruction, and supportive care to address volume depletion and electrolyte disturbances."
1. Identify and Treat the Cause
- Discontinue nephrotoxins (NSAIDs, aminoglycosides, contrast, ACE inhibitors if prerenal)
- Relieve urinary obstruction (Foley catheter, ureteric stent, percutaneous nephrostomy)
- Treat sepsis aggressively (source control, antibiotics, hemodynamic support)
- Treat AIN with steroids when drug-induced and offending agent removed
2. Fluid and Hemodynamic Management
- Prerenal AKI: IV fluids (isotonic crystalloids preferred); target euvolemia
- Avoid fluid overload - associated with worse outcomes in AKI
- Vasopressors in septic shock to maintain MAP ≥65 mmHg
- Balanced crystalloids (e.g., lactated Ringer's) are preferred over normal saline to reduce hyperchloremic acidosis and AKI risk
3. Electrolyte Management
- Hyperkalemia: Dietary restriction, sodium bicarbonate, calcium gluconate (membrane stabilization), insulin/dextrose, kayexalate, dialysis if severe
- Metabolic acidosis: Sodium bicarbonate if pH <7.2 or HCO3 <15
- Hyperphosphatemia: Dietary restriction, phosphate binders
- Hyponatremia/hypervolemia: Fluid restriction
4. Nutritional Support
- Standard caloric needs (20-25 kcal/kg/day non-catabolic; 25-30 in hypercatabolic)
- Protein: 0.8-1.0 g/kg/day (non-dialysis); 1.0-1.5 g/kg/day if on RRT
5. Drug Dosing Adjustments
- Renally cleared drugs must be dose-adjusted (antibiotics, anticoagulants, antivirals)
- Avoid NSAIDs, contrast, aminoglycosides when possible
6. Renal Replacement Therapy (RRT)
Indications (AEIOU):
- Acidosis refractory to medical management
- Electrolyte disturbance (hyperkalemia) not responding to treatment
- Ingestion/intoxication
- Overload (fluid overload refractory to diuretics)
- Uremic symptoms (encephalopathy, pericarditis, coagulopathy, nausea/vomiting)
Modalities:
- Intermittent Hemodialysis (IHD): Hemodynamically stable patients; more efficient solute removal
- Continuous RRT (CRRT): Hemodynamically unstable patients; preferred in ICU
- Peritoneal dialysis: Alternative when vascular access not available
Timing of RRT: Multiple RCTs have shown that in patients with KDIGO Stage 3 AKI, delayed initiation (watchful waiting) showed no increase in mortality compared to early initiation, and patients with delayed dialysis often had earlier diuresis and fewer adverse events. - Goldman-Cecil Medicine, p. 1349
Complications of AKI
| Complication | Mechanism |
|---|---|
| Hyperkalemia | Reduced K+ excretion + tissue breakdown |
| Metabolic acidosis | Reduced H+ excretion, reduced HCO3 reabsorption |
| Hyperphosphatemia | Reduced phosphate excretion |
| Volume overload/pulmonary edema | Reduced urine output |
| Uremia | Accumulation of urea and other uremic toxins |
| Hyponatremia | Water retention exceeding Na retention |
| Anemia | Reduced EPO production |
| Infection | Immune dysregulation, indwelling catheters |
| AKI-CKD progression | ~25-50% of severe AKI survivors develop CKD |
Prognosis and AKI-to-CKD Transition
- About 1/3 of hospitalized patients with AKI resolve within 24 hours, ~60% by 48 hours, and ~70% by 72 hours
- ATN carries a higher risk of CKD progression than non-ATN AKI
- Severe AKI requiring dialysis carries ~40-60% in-hospital mortality in ICU settings
- Each episode of AKI independently increases the risk of CKD, ESRD, cardiovascular events, and all-cause mortality
- Long-term follow-up with nephrology is essential after hospitalized AKI
Prevention
- Identify high-risk patients early (CKD, elderly, diabetic, volume-depleted, septic)
- Minimize nephrotoxin exposure; use N-acetylcysteine + hydration for contrast nephropathy
- Optimize hemodynamics before major surgery
- Use balanced crystalloids rather than normal saline for resuscitation
- Biomarker-guided (TIMP-2 × IGFBP7) protocols may allow preventive care bundles in high-risk surgical patients
Key Sources:
- Goldman-Cecil Medicine International Edition, Ch. 106 - Acute Kidney Injury, p. 1241-1243
- NKF Primer on Kidney Diseases, 8e, Ch. 31 - Acute Kidney Injury Definition, p. 346-348
- Brenner and Rector's The Kidney, 2-Volume Set - AKI Biomarkers and Pathophysiology chapters
- Sabiston Textbook of Surgery, 14e - KDIGO Criteria Table 26.4, p. 449
- Frameworks for Internal Medicine - AKI Categories, p. 448
cause of aki in detail with rational
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
I now have all the material needed for a detailed, well-structured, rational account of AKI causes. Let me compose the full answer.
Causes of AKI - In Detail with Rationale
AKI causes are divided into three anatomic categories, each with a distinct pathophysiologic basis:
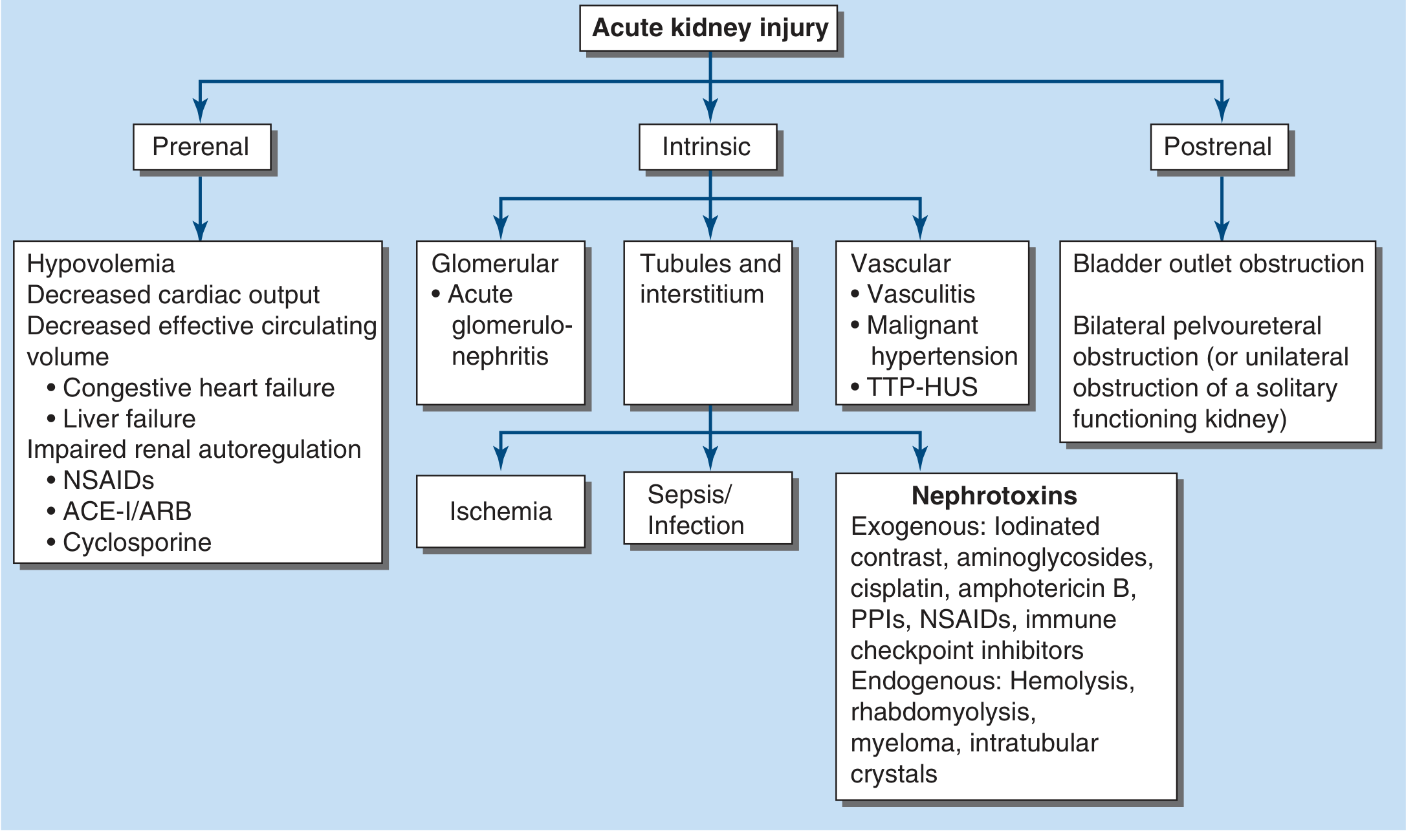
CATEGORY 1: PRERENAL AKI (40-80% of all AKI)
Core Rationale
Prerenal AKI arises from inadequate renal plasma flow and intraglomerular hydrostatic pressure to support normal glomerular filtration - without any structural damage to the kidney itself. The kidney parenchyma is intact; the problem is upstream. Because no tissue is destroyed, it is fully reversible within hours of restoring perfusion. - Harrison's Principles of Internal Medicine, 22e
Normal Autoregulatory Defense (Why Prerenal AKI Has a Threshold Effect)
The kidney defends GFR through three autoregulatory mechanisms:
- Myogenic reflex: Afferent arteriole dilates in response to reduced perfusion pressure
- Prostaglandins (PGI2, PGE2): Vasodilate the afferent arteriole under hypoperfusion
- Angiotensin II: Preferentially vasoconstricts the efferent arteriole to maintain glomerular capillary pressure
- Tubuloglomerular feedback: Macula densa senses low solute delivery → afferent arteriole dilates → maintains GFR
These compensatory responses maintain GFR until mean arterial pressure falls below ~80 mmHg, at which point GFR drops steeply and AKI ensues.
Causes of Prerenal AKI with Rationale
A. True Hypovolemia (Reduced Circulating Blood Volume)
| Cause | Mechanism |
|---|---|
| GI losses (vomiting, diarrhea, NGT drainage) | Fluid lost from the gut → reduced intravascular volume → low renal perfusion pressure |
| Hemorrhage | Direct loss of blood volume → reduced MAP → reduced afferent arteriolar pressure |
| Burns | Massive plasma leak into burn wound (third-space loss) → reduced effective circulating volume |
| Diuretic overuse | Forced sodium and water excretion → reduced intravascular volume |
| Insensible losses (fever, sweating) | Increased free water loss not replaced → hypovolemia |
| Pancreatitis/Peritonitis | Third-space fluid sequestration into inflamed cavity → decreased effective circulating volume |
Rationale: All of these reduce the effective blood volume reaching the kidneys. The reduced afferent arteriolar pressure triggers the renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system. While angiotensin II initially helps by constricting the efferent arteriole, once perfusion falls below the autoregulatory threshold, GFR falls.
B. Reduced Cardiac Output (Effective Arterial Hypovolemia)
| Cause | Mechanism |
|---|---|
| Myocardial infarction | Pump failure → low cardiac output → low renal perfusion pressure |
| Cardiomyopathy / Congestive Heart Failure | Despite fluid overload, the effective arterial volume (volume perceived as filling the vascular tree) is low → RAAS and SNS activation → renal vasoconstriction |
| Valvular disease (severe aortic stenosis, mitral stenosis) | Low forward flow → reduced MAP and renal perfusion |
| Cardiac tamponade/massive PE | Obstructive shock → reduced cardiac output |
Rationale: In heart failure, the kidneys "see" underfilling of the arterial tree (low effective arterial volume) despite total body sodium and water excess. This triggers intense RAAS activation and ADH release. The kidneys retain sodium and water (worsening edema) while GFR falls - a vicious cycle. The term "cardiorenal syndrome" describes this bidirectional interaction. - Rosen's Emergency Medicine
C. Decreased Effective Arterial Volume (Distributive States)
| Cause | Mechanism |
|---|---|
| Liver failure / Cirrhosis | Splanchnic vasodilation (from nitric oxide) → underfilling of systemic circulation → compensatory RAAS/SNS activation → renal vasoconstriction → hepatorenal syndrome |
| Nephrotic syndrome | Profound hypoalbuminemia → low oncotic pressure → fluid escapes to interstitium → reduced effective circulating volume |
| Sepsis (early/distributive) | Massive peripheral vasodilation → profound drop in SVR → decreased effective perfusion pressure despite high cardiac output |
| Anaphylaxis | Extreme vasodilation and capillary leak → acute distributive shock → reduced renal perfusion |
Special note - Hepatorenal Syndrome: This represents the extreme end of cirrhotic prerenal physiology. Intense splanchnic vasodilation causes compensatory renal vasoconstriction via RAAS, ADH, and endothelin. The kidney itself is normal - when transplanted into a healthy recipient, it functions perfectly. It is defined by urine sodium <10 mEq/L, absence of proteinuria, and failure to respond to volume expansion.
D. Impaired Renal Autoregulation (Drug-Induced Prerenal AKI)
This is one of the most clinically important and avoidable causes:
NSAIDs (including COX-2 inhibitors)
- Normal state: Renal prostaglandins (PGI2, PGE2) play a minor role in renal blood flow
- Hypoperfused state: Prostaglandins become critically important for maintaining afferent arteriolar dilation
- NSAIDs block COX-1/COX-2 → prostaglandin synthesis inhibited → afferent vasoconstriction → GFR falls
- Risk is highest in elderly, CKD, CHF, cirrhosis, or volume-depleted patients
ACE Inhibitors / ARBs
- Angiotensin II normally maintains GFR under hypoperfusion by constricting the efferent arteriole
- ACE-I/ARBs block this → efferent arteriole dilates → glomerular capillary hydrostatic pressure drops → GFR falls
- Critical scenario: Bilateral renal artery stenosis - GFR is entirely dependent on efferent vasoconstriction; ACE-I/ARB causes precipitous GFR loss
- Also important: combined NSAID + ACE-I use ("triple whammy" with diuretic) dramatically increases AKI risk
Cyclosporine / Tacrolimus (Calcineurin inhibitors)
- Cause intense afferent arteriolar vasoconstriction → reduced GFR
- This is a hemodynamic (prerenal-type) effect, distinct from their chronic nephrotoxicity
SGLT-2 inhibitors lower intraglomerular pressure by reducing proximal tubule sodium reabsorption, but recent studies show a protective effect against AKI - probably through reduction in hyperfiltration injury and anti-inflammatory effects. - Harrison's Principles, 22e
CATEGORY 2: INTRINSIC (RENAL) AKI
Core Rationale
Intrinsic AKI involves structural damage to the kidney parenchyma - tubules, interstitium, glomeruli, or blood vessels. Unlike prerenal AKI, restoring perfusion does NOT immediately reverse function because cells have been destroyed. Recovery requires cellular regeneration, which takes days to weeks.
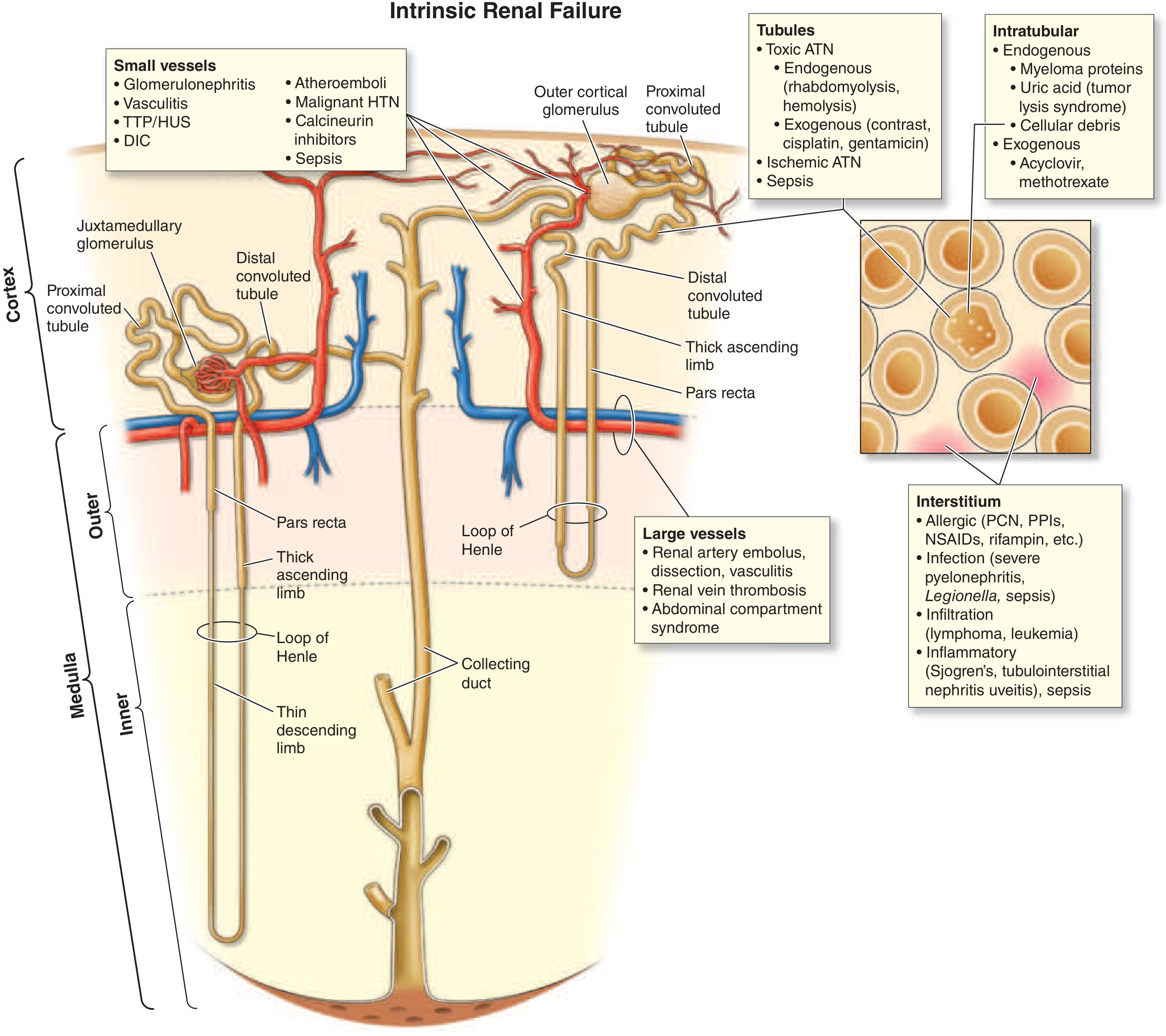
2A: TUBULAR INJURY - Acute Tubular Necrosis (ATN) - 80-90% of Intrinsic AKI
The S3 segment of the proximal tubule (pars recta) and the medullary thick ascending limb are the most vulnerable because:
- They have the highest metabolic demand (ATP-intensive active transport)
- They exist in the most hypoxic zone of the kidney (renal medulla has a naturally low pO2)
- They cannot switch to anaerobic glycolysis under ischemia
i. Ischemic ATN
Causes: Prolonged prerenal azotemia, cardiogenic shock, septic shock, aortic cross-clamping, major surgery, hemorrhagic shock, burns
Pathophysiology - Step by Step:
- ATP depletion → failure of Na-K-ATPase → cells swell, lose polarity
- Cytoskeletal disruption → loss of brush border → tubular cells detach into lumen
- Detached cells + cellular debris form casts → tubular obstruction → raised intratubular pressure → backpressure on glomerulus → GFR falls
- Backleak of filtrate across denuded, leaky tubular epithelium → net fluid absorption is impaired
- Afferent arteriolar vasoconstriction (from tubuloglomerular feedback - macula densa senses increased distal solute delivery due to failed proximal reabsorption) → further reduces GFR
- Inflammatory cascade: Neutrophils and monocytes infiltrate the interstitium → cytokine release (TNF-α, IL-1, IL-6) → endothelial injury → microvascular thrombosis → worsens medullary ischemia
- Reactive oxygen species (ROS) from mitochondrial dysfunction cause lipid peroxidation, cell membrane damage, and apoptosis
"Transient ischemia alone in a normal kidney is usually not sufficient to cause severe AKI, as evidenced by the relatively low risk of severe AKI even after total interruption of renal blood flow during suprarenal aortic clamping or cardiac arrest." Prerenal azotemia and ischemic ATN represent a continuum. - Harrison's Principles, 22e
ii. Sepsis-Associated AKI (most common cause of ATN in ICU)
Pathophysiology:
- Renal vasoconstriction: Activation of RAAS, sympathetic nervous system, vasopressin, and endothelin
- Paradox: In early sepsis, renal blood flow may actually be normal or even high, yet GFR falls - suggesting altered intrarenal microvascular distribution (shunting away from medullary tubules)
- Endothelial damage → increased leukocyte adhesion and migration → microvascular thrombosis → tubular ischemia
- Cytokine storm: TNF-α, IL-1, IL-6, IL-8 → direct tubular toxicity, increased permeability
- Reactive oxygen species from activated neutrophils and macrophages → oxidative tubular damage
- Sepsis may also cause AIN (see below)
iii. Exogenous Nephrotoxins
Radiocontrast Agents (Contrast-Induced AKI)
- Mechanism 1 - Renal medullary ischemia: Contrast causes intense intrarenal vasoconstriction via endothelin, adenosine, and reduced nitric oxide → medullary hypoxia (S3 segment and thick ascending limb most affected)
- Mechanism 2 - Direct tubular cytotoxicity: Contrast agents are directly toxic to tubular epithelial cells via reactive oxygen species and apoptosis
- Timeline: SCr rises at 24-48 hours, peaks at 3-5 days, typically resolves within 1 week
- Risk factors: CKD, diabetes, volume depletion, high contrast volume, heart failure, NSAID/ACE-I use
- Prevention: IV fluid hydration with isotonic saline before and after, minimize contrast dose, use iso-osmolar agents
Aminoglycosides (Gentamicin, Tobramycin, Amikacin)
- Freely filtered at glomerulus → concentrated in proximal tubular cells (via megalin receptor)
- Accumulate in lysosomes → phospholipidosis → lysosomal rupture → mitochondrial dysfunction → cell death
- Non-oliguric AKI in 10-30% of courses, even at therapeutic levels
- Onset: typically 5-7 days into therapy (can appear even after stopping)
- Causes hypomagnesemia (tubular Mg wasting) as an early sign
- Once-daily dosing reduces nephrotoxicity compared to multiple daily doses
Amphotericin B
- Binds ergosterol (and cholesterol) in tubular cell membranes → pores form → tubular cell lysis
- Also causes intense afferent arteriolar vasoconstriction → ischemia
- Causes distal RTA, hypokalemia, hypomagnesemia, and nephrotoxic ATN
Vancomycin
- Tubular injury, especially when trough levels are high
- Can crystallize in tubules causing intratubular obstruction
- Synergistically nephrotoxic with aminoglycosides and piperacillin-tazobactam
Cisplatin and other Platinum Chemotherapeutics
- Enter proximal tubular cells via organic cation transporters
- Form platinum-DNA adducts in tubular cell nuclei → apoptosis and necrosis
- Non-oliguric AKI; also causes hypomagnesemia (tubular Mg wasting), hypokalemia
iv. Endogenous Nephrotoxins
Myoglobin (Rhabdomyolysis)
Causes: Crush injury, prolonged immobilization, seizures, statin toxicity, extreme exertion, heat stroke, drugs (cocaine, ecstasy), hypokalemia, hypothyroidism
Triple mechanism of renal damage:
- Cast formation: Myoglobin precipitates with Tamm-Horsfall protein in acidic, concentrated urine → obstructive casts in distal tubules → tubular obstruction
- Direct proximal tubular cytotoxicity: At pH ≤5.6, myoglobin dissociates into ferriheme + free iron → ferriheme causes lipid peroxidation → oxidative cell death in proximal tubules
- Intrarenal vasoconstriction: Myoglobin scavenges nitric oxide (a potent renal vasodilator) → net vasoconstrictive effect; also activates endothelin-1, thromboxane A2, and RAAS
All three mechanisms are worsened by volume depletion and aciduria - hence the rationale for aggressive isotonic saline (± bicarbonate to alkalinize urine) as treatment. - Rosen's Emergency Medicine
Hemoglobin (Hemolysis - Pigment Nephropathy)
- Mechanism is analogous to myoglobin-induced ATN
- Causes: Transfusion reactions, G6PD deficiency, TTP/HUS, malaria, snake envenomation
- Hemoglobin cast formation in distal tubules + proximal tubular direct toxicity via heme iron
Uric Acid Crystals (Acute Urate Nephropathy)
- Context: Tumor Lysis Syndrome (after cytotoxic therapy for high-burden tumors - lymphoma, leukemia) or spontaneous (Burkitt's, ALL)
- Massive cell death → huge purine load → uric acid overproduction → uric acid precipitates in collecting ducts and distal tubules (poorly soluble at acidic pH)
- Causes: tubular obstruction → anuria; also intrarenal vasoconstriction
- Treatment: rasburicase, allopurinol, IV hydration, urine alkalinization
Myeloma Cast Nephropathy ("Myeloma Kidney")
- Light chains (Bence Jones proteins) filtered at glomerulus → co-precipitate with Tamm-Horsfall protein in distal tubules → obstruct tubules
- Light chains also directly toxic to proximal tubule cells (endocytosis → lysosomal injury)
- Volume depletion, contrast, hypercalcemia, and NSAIDs dramatically worsen myeloma AKI
Calcium Oxalate Crystals
- Ethylene glycol ingestion → oxalic acid → calcium oxalate crystals → tubular obstruction
- Also in primary oxaluria, after jejunoileal bypass
2B: ACUTE INTERSTITIAL NEPHRITIS (AIN) - 5-10% of Intrinsic AKI
Core Rationale: An immune-mediated inflammatory reaction in the renal interstitium, not primarily the tubules. Pathologically shows interstitial edema and inflammatory infiltrate (T lymphocytes, macrophages, ± eosinophils) with relative preservation of glomeruli.
Causes:
1. Drug-Induced (most common ~75%)
- Mechanism: Drugs act as haptens or directly trigger a T-cell-mediated delayed hypersensitivity reaction against tubular antigens
- Key drugs:
| Drug Class | Examples |
|---|---|
| Beta-lactam antibiotics | Penicillin, methicillin, cephalosporins |
| Sulfonamides | TMP-SMX |
| Fluoroquinolones | Ciprofloxacin |
| Rifampin | - |
| NSAIDs | Any NSAID (can also cause nephrotic syndrome simultaneously) |
| Proton pump inhibitors | Omeprazole, pantoprazole (now a leading cause) |
| Diuretics | Furosemide, thiazides |
| Checkpoint inhibitors | Ipilimumab, nivolumab, pembrolizumab (~5% of treated patients) |
| Allopurinol | - |
Classic triad (present in only ~1/3 of cases): Fever + rash + eosinophilia - absence does NOT exclude AIN.
2. Infection-Associated AIN
- Mechanism: Direct infection or immune-complex deposition
- Legionella pneumophila, Leptospira, Streptococcus, Mycobacterium tuberculosis, CMV, EBV, SARS-CoV-2, Hantavirus
3. Autoimmune / Systemic Disease
- Sjogren's syndrome, SLE, sarcoidosis (granulomatous AIN), tubulointerstitial nephritis-uveitis (TINU) syndrome, IgG4-related disease
2C: ACUTE GLOMERULONEPHRITIS - <5% of Intrinsic AKI
Core Rationale: Immune-mediated inflammation directly in the glomerulus → reduced filtration surface area + increased glomerular capillary permeability. GFR falls because glomerular inflammation causes:
- Proliferation of mesangial, endothelial, or epithelial cells → physically narrows the capillary lumen
- Fibrin deposition → occludes capillary loops
- Inflammatory cells → release cytokines/ROS damaging the filtration barrier
Rapidly Progressive GN (RPGN) - most severe form:
| Type | Mechanism | Cause |
|---|---|---|
| Type I - Anti-GBM | Antibodies vs. collagen IV in GBM → linear IgG deposits | Goodpasture's syndrome (lung + kidney) |
| Type II - Immune complex | Immune complex deposition → complement activation → inflammation | Post-streptococcal GN, lupus nephritis, IgA nephropathy (cresentic), endocarditis |
| Type III - Pauci-immune (ANCA) | ANCA-mediated neutrophil activation → vessel wall destruction | GPA (Wegener's), MPA, EGPA (Churg-Strauss) |
Clinical clues: Hematuria with red cell casts, proteinuria (often 1-3 g/day), hypertension, edema. Absence of red cell casts, hematuria, and significant proteinuria effectively excludes GN.
2D: VASCULAR CAUSES - Large and Small Vessel Disease
Large Vessel Disease
| Cause | Mechanism |
|---|---|
| Renal artery occlusion (thromboembolism, dissection, vasculitis) | Abrupt cessation of arterial inflow → cortical ischemia/infarction |
| Renal vein thrombosis | Venous congestion → reduced net filtration pressure → AKI (particularly in nephrotic syndrome, hypercoagulable states) |
| Abdominal compartment syndrome | Raised intra-abdominal pressure → renal vein and inferior vena cava compression → venous hypertension → reduced perfusion pressure |
| Aortic dissection | Involvement of renal artery ostia → ischemic AKI |
Small Vessel and Microvascular Disease
| Cause | Mechanism |
|---|---|
| TTP (Thrombotic Thrombocytopenic Purpura) | ADAMTS13 deficiency → uncleaved vWF multimers → platelet aggregation → microthrombi in glomerular capillaries → microangiopathic hemolytic anemia + AKI |
| HUS (Hemolytic Uremic Syndrome) | Shiga toxin (E. coli O157:H7) → endothelial injury in renal microvasculature → microthrombi in glomeruli → triad: hemolytic anemia + thrombocytopenia + AKI (predominant in children) |
| Malignant Hypertension | Extremely high BP → fibrinoid necrosis of arterioles → ischemic glomerular injury + tubular ischemia |
| Atheroembolic AKI | Cholesterol crystals embolize (after aortic catheterization or anticoagulation) → lodge in small renal arteries → local inflammation + ischemia; subacute onset; livedo reticularis, eosinophilia |
| Scleroderma Renal Crisis | Intense arteriolar spasm + endothelial proliferation → critical renal ischemia → malignant HTN + AKI |
| HELLP Syndrome / Preeclampsia | Endothelial dysfunction + microangiopathy in pregnancy → glomerular endotheliosis + platelet thrombi |
| Vasculitis (ANCA, PAN) | Necrotizing inflammation of arterioles → fibrinoid necrosis → ischemic glomerular injury |
CATEGORY 3: POSTRENAL AKI (5-10% of all AKI)
Core Rationale
Obstruction at any level of the urinary tract causes a retrograde rise in intratubular pressure that opposes glomerular filtration. Initially afferent arteriole dilates (hyperemia), but within hours, intrarenal vasoconstriction from angiotensin II, thromboxane A2, and vasopressin causes GFR to fall. The key insight: obstruction must be bilateral (or unilateral in a solitary functioning kidney) to cause AKI.
Pathophysiology of Obstruction
- Acute obstruction → ↑ intratubular pressure → opposes net filtration pressure → GFR falls
- Phase 1 (0-2 hrs): Compensatory afferent arteriolar dilation (prostaglandin-mediated) → increased renal blood flow
- Phase 2 (2-5 hrs): Efferent arteriolar vasodilation → reduced filtration pressure
- Phase 3 (>5 hrs): Afferent arteriolar vasoconstriction (angiotensin II, thromboxane A2, endothelin) → marked reduction in RBF and GFR
- Prolonged obstruction → tubular atrophy + interstitial fibrosis → permanent CKD
Causes by Level
Intrarenal (Collecting Duct Level)
- Crystal precipitation: Uric acid (tumor lysis), calcium oxalate (ethylene glycol), phosphate (bowel prep), acyclovir, sulfonamides, methotrexate, indinavir
- Myeloma proteins (see above)
Ureteral (Bilateral or Solitary Kidney)
- Nephrolithiasis (most common cause of bilateral ureteral obstruction in young patients)
- Retroperitoneal fibrosis (idiopathic or secondary to drugs - methysergide, beta-blockers; associated with AAA)
- Pelvic/retroperitoneal malignancy (cervical, bladder, prostate, lymphoma, metastases)
- Sloughed renal papillae (diabetes, analgesic nephropathy, sickle cell disease)
- Surgical ligation of ureter
- Blood clots
Bladder
- Benign prostatic hypertrophy - most common cause of postrenal AKI in older men
- Prostate cancer
- Neurogenic bladder (diabetic autonomic neuropathy, spinal cord injury, MS)
- Anticholinergic drugs (tricyclics, antihistamines, antipsychotics) → bladder atony
- Bladder carcinoma
- Blood clots, bladder stones
Urethra
- Urethral stricture (post-infectious, post-traumatic)
- Phimosis (in children)
- Obstructed urinary catheter (most preventable cause in hospital)
"Obstruction is an eminently reversible cause of AKI and should be considered in every patient with newly discovered azotemia." - Rosen's Emergency Medicine
A post-obstructive diuresis can occur after relief - the kidneys excrete retained salt, water, and urea; patients need careful IV fluid replacement to avoid volume depletion worsening AKI.
Summary Table: Distinguishing the Three Categories
| Feature | Prerenal | Intrinsic (ATN) | Postrenal |
|---|---|---|---|
| BUN:Cr | >20:1 | ~10-15:1 | Variable |
| FENa | <1% | >2% | Variable |
| Urine Na | <10 mEq/L | >40 mEq/L | Variable |
| Urine osmolality | >500 | <350 | Variable |
| Urine sediment | Normal / hyaline casts | Muddy brown granular casts, tubular cells | Normal or RBC casts if stone |
| Response to fluids | Rapid reversal | No immediate reversal | No reversal until obstruction relieved |
| Kidney size on US | Normal | Normal | Bilateral hydronephrosis |
| Biomarkers (NGAL, KIM-1) | Low | Very elevated | Low-moderate |
Key Sources:
- Harrison's Principles of Internal Medicine, 22e (2025) - Chapter 321, Prerenal Azotemia and Ischemia/Nephrotoxin-Associated AKI
- Rosen's Emergency Medicine - Chapters 83 and 116 (Rhabdomyolysis)
- Goldman-Cecil Medicine International Edition - Chapter 106, Intrinsic AKI
- Frameworks for Internal Medicine - Chapter 33, AKI Categories
- Smith and Tanagho's General Urology, 19e - Postrenal Kidney Injury
This is a shared conversation. Sign in to Orris to start your own chat.