Acute flaccid paralysis case presentation for MD Pediatrics
Reading File
Finding Sources
Searching the Web
acute flaccid paralysis case presentation MD Pediatrics clinical approach 2024
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
I now have comprehensive information to construct a full MD Pediatrics case presentation on Acute Flaccid Paralysis. Let me compile it.
Acute Flaccid Paralysis (AFP) - Case Presentation for MD Pediatrics
CASE VIGNETTE
Chief Complaint: A 4-year-old boy presents with sudden-onset weakness of the left lower limb of 3 days' duration.
History of Present Illness:
- 3 days ago, parents noticed the child was limping and unable to lift his left leg while walking.
- 10 days prior, he had fever (38.5°C), runny nose, and mild cough lasting 5 days (resolved spontaneously).
- No head trauma. No rash. No back pain initially, though dull aching in the left thigh was noted at onset of weakness.
- Bowel and bladder dysfunction noted (urinary retention for 12 hours before admission).
- No history of recent diarrhea. No animal bites.
- Vaccination history: Received OPV at birth, 6, 10, 14 weeks, and at 16-18 months. Last dose 2.5 years ago. No recent travel to polio-endemic areas.
Past History: Uneventful. No prior neurological illness.
Family History: Non-contributory.
PHYSICAL EXAMINATION
Vitals: Temp 37.2°C | HR 98/min | RR 22/min | SpO2 98% on room air
General Examination: Alert, mildly irritable. No meningismus. No lymphadenopathy.
Neurological Examination:
| Parameter | Left Lower Limb | Right Lower Limb | Upper Limbs |
|---|---|---|---|
| Tone | Decreased (hypotonia) | Normal | Normal |
| Power (MRC) | 1/5 (proximal) | 5/5 | 5/5 |
| Deep tendon reflexes | Absent | Present 2+ | 2+ bilaterally |
| Plantar response | Absent/equivocal | Flexor | Flexor |
| Sensation | Intact | Intact | Intact |
- Cranial nerves: Intact
- Spine: No tenderness, no gibbus
- Bladder: Distended on palpation
Key clinical findings: Acute, asymmetric, flaccid weakness with areflexia, preserved sensation, and bladder involvement in a febrile prodrome context = classic Lower Motor Neuron (LMN) syndrome.
DEFINITION
AFP is defined as acute onset of flaccid (reduced tone) paralysis in any child under 15 years of age, or at any age when poliomyelitis is suspected. It is a WHO surveillance indicator for poliomyelitis eradication. A country's surveillance system must detect at least 1 AFP case per 100,000 children under 15 annually, even in the absence of polio. - Park's Textbook of Preventive and Social Medicine
ETIOLOGY / CLASSIFICATION
AFP is a clinical syndrome with many causes across different anatomical levels:
Anterior Horn Cell (Spinal/Brainstem)
- Poliovirus (types 1, 2, 3) - most historically important
- Non-polio enteroviruses: EV-D68 (major cause of biennial AFM outbreaks in USA since 2014), EV-A71 (Asia-Pacific; associated with HFMD), CV-A7
- Arboviruses: West Nile virus, Japanese Encephalitis virus, Tick-borne encephalitis virus
- Acute Flaccid Myelitis (AFM)
Peripheral Nerve (Polyradiculoneuropathy)
- Guillain-Barré Syndrome (AIDP, AMAN, AMSAN) - now the leading cause of AFP in vaccinated populations
- Diphtheritic polyneuropathy
- Tick paralysis
Neuromuscular Junction
- Botulism (descending paralysis + cranial nerve palsies)
- Myasthenia gravis
Muscle
- Hypokalemic periodic paralysis
- Viral myositis
Spinal Cord
- Transverse myelitis (associated with ADEM, NMO spectrum)
- Spinal cord compression
INVESTIGATIONS
Mandatory (WHO AFP Protocol)
- Two stool samples within 14 days of onset, taken 24-48 hours apart, stored at 4-8°C, sent to WHO-accredited lab within 72 hours - for poliovirus isolation and typing (wild vs. vaccine-derived)
Workup for Etiology
| Investigation | Rationale |
|---|---|
| CBC, ESR, CRP | Systemic infection/inflammation |
| CSF analysis | Distinguish polio, GBS, AFM, TM |
| Stool culture/PCR | Enterovirus, poliovirus |
| Nasopharyngeal swab PCR | EV-D68 (respiratory predilection) |
| Nerve conduction study (NCS) + EMG | Essential: LMN vs. demyelinating; helps confirm GBS |
| MRI spine (T2-weighted) | AFM - T2 hyperintensity in anterior horn (gray matter); TM - central T2 lesion |
| Serum electrolytes (K+) | Hypokalemic paralysis |
| Anti-ganglioside antibodies (anti-GM1, anti-GQ1b) | GBS subtypes |
CSF Findings - Distinguishing Features
| Condition | Cells | Protein | Glucose |
|---|---|---|---|
| Poliovirus / AFM | Lymphocytic pleocytosis (<100/μL) | Mildly elevated | Normal |
| GBS (AIDP) | <10 cells/μL | Elevated (albuminocytological dissociation) | Normal |
| Transverse Myelitis | Variable pleocytosis | Elevated | Normal |
| Normal | 0-5 lymphocytes | 15-45 mg/dL | 50-75% serum |
Source: Goldman-Cecil Medicine, Bradley & Daroff's Neurology in Clinical Practice
DIFFERENTIAL DIAGNOSIS - Distinguishing Features
Poliomyelitis / AFM
- Prodrome: fever + URTI/gastroenteritis
- Onset: rapid, hours to days
- Pattern: asymmetric, proximal > distal
- Sensory: intact
- Reflexes: absent in affected limb
- CSF: mild pleocytosis
- MRI: T2 hyperintensity in anterior horn cells - Goldman-Cecil Medicine
Guillain-Barré Syndrome
- Prodrome: URTI or gastroenteritis (1-4 weeks prior; Campylobacter jejuni most common trigger)
- Onset: ascending, over days to 4 weeks
- Pattern: symmetric, distal to proximal, ascending
- Sensory: mild distal sensory loss/paresthesias
- Cranial nerve: facial diplegia in 45-75% of cases
- Autonomic dysfunction: 65% (BP instability, arrhythmias, urinary retention)
- CSF: albuminocytological dissociation (elevated protein, <10 cells)
- NCS: conduction slowing/block (AIDP) or axonal loss (AMAN/AMSAN)
- Source: Bradley & Daroff's Neurology in Clinical Practice
Transverse Myelitis
- Both motor and sensory involvement below the lesion
- Sensory level present
- Upper motor signs at/above lesion, lower motor below
- Bladder/bowel involvement common
- MRI: central T2 lesion spanning the cord
Botulism
- Descending flaccid paralysis (cranial nerves first)
- Fixed dilated pupils, diplopia, dysphagia
- Fully conscious patient
- No fever
- Foodborne exposure or wound source
- CSF: normal - Harrison's Principles of Internal Medicine 22e
Tick Paralysis
- Ascending flaccid paralysis without sensory loss
- No fever
- History of tick attachment (often behind ear/scalp)
- Resolves on tick removal - Tintinalli's Emergency Medicine
CASE DIAGNOSIS - Working Through the Differential
In this case:
- Asymmetric flaccid monoplegia -> favors polio/AFM over GBS (which is symmetric)
- Respiratory prodrome 10 days prior -> consistent with EV-D68-associated AFM
- Preserved sensation -> anterior horn cell disease, not transverse myelitis
- Bladder involvement -> occurs in both AFM and TM
- Vaccination history: OPV given, but >2 years since last dose; wild poliovirus or VDPV must be excluded
Most likely diagnosis: Acute Flaccid Myelitis (AFM) secondary to Enterovirus D68
Must exclude: Vaccine-derived poliovirus (VDPV) / Wild poliovirus - stool samples mandatory per WHO protocol
MANAGEMENT
Acute Phase
- Hospitalization - monitor for respiratory compromise (bulbar involvement, respiratory muscle weakness)
- Respiratory support - if SpO2 falls, PaO2 <60 or PaCO2 >50 mmHg: NIPPV or mechanical ventilation
- Neurological monitoring - serial power charting every 4-6 hours
- Pain control for muscle spasms
- Bladder care - intermittent catheterization for urinary retention
- Nutritional support - NG feeds if dysphagia
Specific Therapy
- No proven specific therapy for polio or AFM (EV-D68)
- For GBS (if diagnosis shifts): IVIG (2 g/kg over 2-5 days) or Plasmapheresis - equally effective
- For Transverse Myelitis: IV methylprednisolone 30 mg/kg/day (max 1 g/day) x 3-5 days
- Corticosteroids are NOT indicated in GBS
Rehabilitation
- Physiotherapy should begin early to prevent contractures and maintain range of motion
- Greatest recovery gains occur in the first 6 months of convalescence
- Low-impact exercise, orthoses as needed - Goldman-Cecil Medicine
PROGNOSIS
| Condition | Prognosis |
|---|---|
| Poliovirus (spinal) | Mortality ~5%; 2/3 have residual paresis; deformities with time |
| AFM (EV-D68) | ~75% limited or no improvement at 30 days; partial recovery in 25% |
| AFM (EV-A71) | 93% recovery of neurological disease |
| GBS | Mortality 1-5% with modern ICU care; most recover significantly |
SURVEILLANCE AND PUBLIC HEALTH (AFP Reporting)
All children <15 years with AFP must be reported within 24 hours of detection. Two stool samples must be collected within 14 days of paralysis onset, 24-48 hours apart, and dispatched to a WHO-accredited polio laboratory. This surveillance is the backbone of the Global Polio Eradication Initiative. - Park's Textbook of Preventive and Social Medicine
AFP rate target: At least 2 non-polio AFP cases per 100,000 children under 15 per year (indicator of surveillance sensitivity).
SUMMARY TABLE - AFP Causes at a Glance
| Feature | Polio/AFM | GBS | TM | Botulism | Tick Paralysis |
|---|---|---|---|---|---|
| Distribution | Asymmetric | Symmetric ascending | Para/tetraplegia | Descending | Ascending |
| Sensory | Absent | Mild distal | Sensory level | Absent | Absent |
| Reflexes | Absent (affected limb) | Absent/reduced | Variable | Absent | Absent |
| Fever prodrome | Yes | No (prior URTI/GI) | Variable | No | No |
| CSF protein | Mild ↑ | Marked ↑ (albuminocytological dissociation) | Mild ↑ | Normal | Normal |
| CSF cells | Mild pleocytosis | <10 | Variable | Normal | Normal |
| MRI | Anterior horn T2 | Normal or nerve root enhancement | Central cord T2 | Normal | Normal |
| Treatment | Supportive | IVIG / Plasmapheresis | IV steroids | Antitoxin | Remove tick |
References:
- Goldman-Cecil Medicine International Edition - Acute Flaccid Paralysis/Myelitis
- Bradley & Daroff's Neurology in Clinical Practice - Acute Poliomyelitis and Other Viral AFP; Guillain-Barré Syndrome
- Park's Textbook of Preventive and Social Medicine - AFP Surveillance
- Localization in Clinical Neurology, 8e - Anterior Horn Cell Syndromes
- Harrison's Principles of Internal Medicine 22e (2025) - Botulism
- Tintinalli's Emergency Medicine - Tick Paralysis
- WHO Global AFP Guidelines 2023
For GBS case presentation
Finding Sources
Searching the Web
Guillain Barre syndrome pediatric case presentation MD Pediatrics 2024 clinical features management
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Excellent - I have everything needed for a complete, exam-ready GBS case presentation.
Guillain-Barré Syndrome (GBS) - Case Presentation for MD Pediatrics
CASE VIGNETTE
Chief Complaint: A 7-year-old girl presents with progressive weakness of both lower limbs for 5 days, now spreading to the upper limbs.
History of Present Illness:
- 5 days ago, parents noticed she was reluctant to walk and complained of aching pain in both thighs and lower back. Initially thought to be "muscle soreness."
- Weakness was initially in the legs (noticed as a clumsy gait), progressively ascending to involve the thighs, then the trunk, and now she cannot raise her arms above her head.
- She cannot stand unassisted since yesterday.
- History of loose stools and low-grade fever 3 weeks prior, lasting 4-5 days, which resolved spontaneously (no treatment sought).
- No bladder or bowel dysfunction at present. No headache. No visual symptoms.
- Vaccination history: Up to date including OPV, no recent immunization.
- No family history of neurological disease. No toxin/animal exposure.
Past Medical History: Unremarkable.
PHYSICAL EXAMINATION
Vitals: HR 110/min | RR 26/min | BP 108/70 mmHg | SpO2 96% on room air | Temp afebrile
General: Alert, anxious-appearing child. Appears uncomfortable. No meningismus.
Cranial Nerve Examination:
- Bilateral mild facial weakness (inability to fully close eyes, flat nasolabial folds bilaterally)
- Gag reflex: reduced
- Other cranial nerves intact
Motor Examination:
| Parameter | Lower Limbs | Upper Limbs | Trunk |
|---|---|---|---|
| Tone | Decreased (diffuse hypotonia) | Mildly decreased | Reduced axial tone |
| Power (MRC) | 2/5 (proximal + distal) | 3/5 | Weak neck flexors |
| Deep tendon reflexes | Absent bilaterally | Absent bilaterally | - |
| Plantar | Absent | - | - |
Sensory Examination:
- Distal diminution of vibration sense in both lower limbs
- Mild glove-and-stocking type sensory loss to light touch
- No sensory level
Autonomic Signs:
- Tachycardia (HR 110) without fever
- Blood pressure lability (noted 140/90 to 90/60 on serial checks)
- Mild diaphoresis
Respiratory:
- Paradoxical breathing noted on careful inspection
- FVC measured at bedside: approximately 18 mL/kg (concerning)
Key clinical picture: Symmetric ascending flaccid tetraparesis with areflexia + bifacial palsy + distal sensory impairment + autonomic instability after a prodromal illness = Classic AIDP (GBS)
DEFINITION AND EPIDEMIOLOGY
GBS is an acute immune-mediated polyradiculoneuropathy characterized by rapidly ascending flaccid weakness, areflexia, and in its classic form, albuminocytological dissociation in the CSF. It is the leading cause of acute flaccid paralysis in vaccinated populations and the most common cause of acute neuromuscular paralysis in children. - Bradley & Daroff's Neurology in Clinical Practice
- Incidence: ~1.8 per 100,000 population/year; ~0.34-0.6 per 100,000 in children
- Sex ratio: Males > females (1.5:1)
- Age: Any age; incidence increases with age (0.8/100,000 in <18 yrs; 3.2/100,000 in >60 yrs)
- Mortality: Reduced from 33% (pre-ICU era) to 1-5% with modern critical care
PATHOPHYSIOLOGY
GBS is an autoimmune attack on peripheral nerve myelin and/or axons, triggered by molecular mimicry from a preceding infection.
Trigger: A preceding infection (1-4 weeks prior) stimulates antibody production that cross-reacts with ganglioside epitopes on peripheral nerve components.
Key triggers:
- Campylobacter jejuni (most common - especially for AMAN subtype; 76% of AMAN cases in China)
- Cytomegalovirus (CMV)
- Epstein-Barr virus (EBV)
- Mycoplasma pneumoniae
- Zika virus, SARS-CoV-2
- Rarely: post-vaccination
Mechanism (AIDP): T-cell and antibody-mediated multifocal inflammatory demyelination of spinal roots and peripheral nerves → conduction block → clinical weakness
Mechanism (AMAN): Anti-GM1/anti-GD1a antibodies bind axolemma at nodes of Ranvier → complement activation (C3d, C5b-9) → macrophage invasion → wallerian-like axonal degeneration - Bradley & Daroff
CLASSIFICATION / SUBTYPES
BOX: GBS Subtypes (Asbury & Cornblath, 1990)
Common Subtypes:
| Subtype | Key Features | EMG/NCS |
|---|---|---|
| AIDP (Acute Inflammatory Demyelinating Polyradiculoneuropathy) | Classic ascending symmetric weakness; most common in West | Conduction slowing, conduction blocks, prolonged F-waves |
| AMAN (Acute Motor Axonal Neuropathy) | Pure motor, no sensory loss; common in Asia; anti-GM1/GD1a Ab | Reduced CMAP, normal SNAP, normal conduction velocity |
| AMSAN (Acute Motor-Sensory Axonal Neuropathy) | Fulminant; severe quadriparesis; poor recovery | Reduced/absent CMAP and SNAP |
Rare Variants:
- Miller-Fisher Syndrome (MFS): Triad of ophthalmoplegia + gait/truncal ataxia + areflexia; anti-GQ1b antibodies; 5-6% of GBS in West, up to 18% in Taiwan - Goldman-Cecil Medicine
- Facial diplegia with paresthesias
- Pharyngeal-cervical-brachial variant
- Acute pandysautonomia
- Paraparetic variant
DIAGNOSTIC CRITERIA (Asbury & Cornblath, 1990)
Features REQUIRED for Diagnosis:
- Progressive weakness of both legs and arms
- Areflexia or hyporeflexia
Clinical Features SUPPORTIVE of Diagnosis:
- Progression over days to 4 weeks
- Relative symmetry of symptoms and signs
- Mild sensory symptoms or signs
- Bifacial palsies
- Autonomic dysfunction
- Absence of fever at onset of weakness
- Recovery beginning 2-4 weeks after progression ceases
Laboratory Features SUPPORTIVE of Diagnosis:
- Elevated CSF protein with <10 cells/μL (albuminocytological dissociation)
- Electrodiagnostic features of nerve conduction slowing or block
Features that CAST DOUBT on diagnosis:
- Marked, persistent asymmetry of weakness
- Persistent bladder/bowel dysfunction (or present at onset)
- CSF mononuclear cells >50/μL
- Sharp sensory level
Features that RULE OUT diagnosis:
- History of hexacarbon abuse
- Abnormal porphyrin metabolism (acute intermittent porphyria)
- Recent diphtheria
INVESTIGATIONS
1. CSF Analysis (Lumbar Puncture)
- Classic finding: Albuminocytological dissociation
- Protein: elevated (>45 mg/dL; can rise to >1000 mg/dL)
- Cells: <10 cells/μL (acellular or near-acellular)
- Glucose: normal
- Present in 50-70% in first week, rises to >90% by week 2-3
- Note: LP can be normal early - repeat in 1 week if clinical suspicion persists
2. Nerve Conduction Studies (NCS) + EMG
The single most informative investigation for subtyping GBS.
| NCS Finding | Interpretation |
|---|---|
| Prolonged distal latencies | Distal demyelination |
| Reduced conduction velocity (<70% LLN) | Demyelination (AIDP) |
| Conduction blocks | Proximal demyelination |
| Prolonged/absent F-waves | Proximal root involvement |
| Reduced/absent CMAP with normal SNAP | AMAN |
| Reduced CMAP + absent SNAP | AMSAN |
3. Anti-ganglioside Antibodies
| Antibody | Association |
|---|---|
| Anti-GM1 | AMAN, AIDP |
| Anti-GD1a | AMAN |
| Anti-GQ1b | Miller-Fisher syndrome (>90% sensitive) |
| Anti-GD1b | AMSAN, sensory GBS |
4. MRI Spine with Contrast
- Nerve root enhancement on T1 gadolinium (not mandatory, but helps in equivocal cases)
- Useful to exclude cord compression/transverse myelitis
5. Respiratory Monitoring (CRITICAL)
- Forced Vital Capacity (FVC) every 4-6 hours during progressive phase
- "20-30-40 Rule":
- FVC < 20 mL/kg
- Maximal inspiratory pressure < 30 cm H₂O
- Maximal expiratory pressure < 40 cm H₂O
- → Any one of these: transfer to ICU, consider intubation
6. Other
- CBC, CMP, electrolytes (hyponatremia = poor prognostic marker)
- Stool culture/PCR for C. jejuni
- Serology: CMV, EBV, Mycoplasma
- ECG (cardiac arrhythmias from autonomic dysfunction)
- Continuous BP monitoring
DIFFERENTIAL DIAGNOSIS
| Condition | Key Distinguishing Features |
|---|---|
| Acute Flaccid Myelitis / Polio | Asymmetric, no sensory loss, pleocytosis in CSF, MRI shows anterior horn T2 changes |
| Transverse Myelitis | Sensory level, upper motor signs above lesion, bladder/bowel at onset, central cord T2 on MRI |
| Botulism | Descending paralysis, dilated pupils, fully conscious, cranial nerves first, normal CSF |
| Myasthenia Gravis | Fatigable weakness, ptosis, diplopia, normal reflexes, repetitive nerve stim decremental |
| Tick Paralysis | Ascending flaccid, no sensory loss, tick found (often occipital/hairline), resolves on removal |
| Hypokalemic Periodic Paralysis | Episodic, normal reflexes between attacks, low K+ |
| Acute Intermittent Porphyria | Abdominal pain, psychiatric features, urine porphyrins elevated |
| Spinal Cord Compression | UMN signs, focal back pain, sensory level, bladder early |
| Diphtheritic Neuropathy | History of pharyngitis, palatal palsy first, no vaccination |
| Critical Illness Polyneuropathy | ICU setting, sepsis, axonal NCS, normal CSF |
MANAGEMENT
A. Emergency Assessment - Respiratory First
The single most important immediate action is to assess respiratory status. Respiratory failure develops in up to 30% of GBS patients.
Predictors of impending respiratory failure (Sharshar rule - "can't lift head, short duration, low FVC"):
- Rapid disease progression (onset to admission <7 days)
- Inability to lift head from bed
- Inability to lift elbows
- Bulbar weakness
- FVC < 60% of predicted
- When all three factors present: 85% probability of requiring ventilation
Elective intubation indications:
- FVC < 12-15 mL/kg (or <18 mL/kg with severe bulbar weakness)
- PaO2 < 70 mmHg on room air
- PaCO2 rising
- Rapidly worsening hypoxia
B. ICU Admission Indications
- Any of the above respiratory signs
- Autonomic instability (BP lability, cardiac arrhythmias)
- Bulbar dysfunction
- Rapid progression
C. Monitoring (Serial every 4-6 hours)
- FVC, negative inspiratory pressure
- Pulse oximetry (especially overnight)
- Continuous ECG and BP monitoring
- Bulbar function / ability to handle secretions
- Motor power charting (MRC scale)
D. Specific Immunotherapy
Both options are equally efficacious and should NOT be combined (no additive benefit).
First choice - IVIG:
- Dose: 0.4 g/kg/day × 5 days (total: 2 g/kg)
- OR 1 g/kg/day × 2 days
- Preferred due to ease of administration and better compliance
- Administer if symptoms <30 days, patient bedbound or worse
- Mechanism: anti-idiotype antibodies, Fc receptor blockade, complement inhibition
Alternative - Plasma Exchange (Plasmapheresis):
- Dose: 200-250 mL plasma/kg over 5 sessions (5 sessions over 7-10 days)
- Equally effective as IVIG
- Less preferred in children due to venous access requirements
- Administer if symptoms <30 days
- More effective if initiated within 7 days of onset
Important:
- Corticosteroids are NOT beneficial in GBS (may worsen outcome)
- Methylprednisolone + IVIG shows slight initial advantage but no long-term benefit over IVIG alone - Goldman-Cecil Medicine
- Combined IVIG + plasma exchange: no additional benefit
- Repeat treatment: generally not recommended
E. Supportive Care
| System | Intervention |
|---|---|
| Pain | Back/radicular pain in ~70%; NSAIDs first; opioids if severe; gabapentin/carbamazepine for neuropathic pain |
| DVT prophylaxis | Subcutaneous LMWH + compression stockings in bedbound patients |
| Nutrition | NG feeds if bulbar palsy; monitor for aspiration |
| Bladder | Intermittent catheterization for urinary retention |
| Skin | 2-hourly turning to prevent pressure ulcers |
| Eyes | Lubricating drops + eye patching for facial nerve palsy |
| Autonomic | Antihypertensives and vasopressors with EXTREME caution (hypotension risk); tracheal suction can trigger vagal bradycardia |
| Bowel | Early management of constipation; prevent ileus |
F. Rehabilitation
- Physiotherapy initiated as soon as clinically stable
- Range-of-motion exercises to prevent contractures
- Progressive ambulation with assistance
- Recovery typically requires months to years - Harriet Lane Handbook
MANAGEMENT FLOWCHART
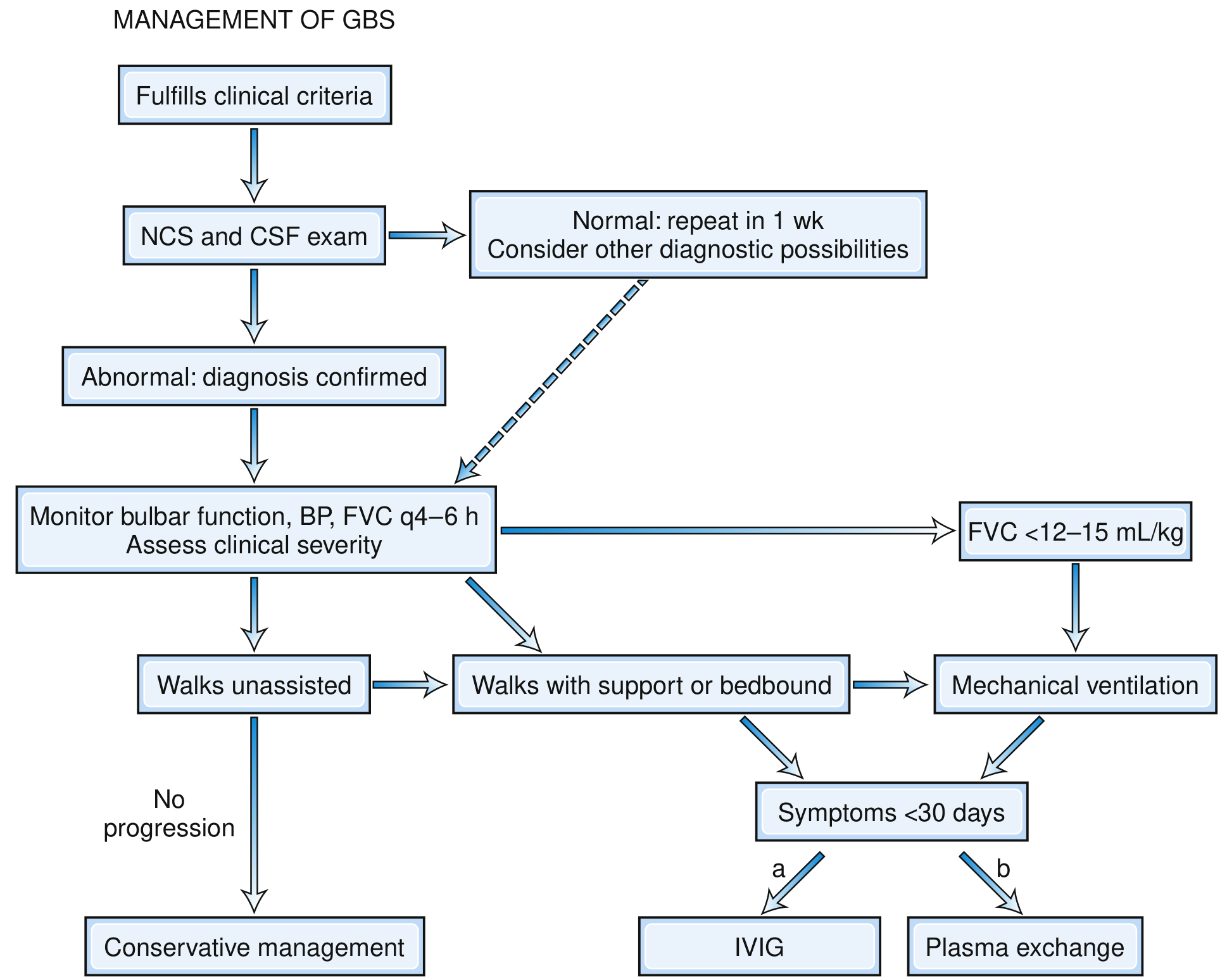
Fig: Decision-Making Pathway in Management of GBS showing that both IVIG and plasma exchange are equally efficacious. IVIG is preferred. Intubation when FVC <12-15 mL/kg. - Bradley & Daroff's Neurology in Clinical Practice
COURSE AND PROGNOSIS
Phases of GBS:
- Progressive phase: Days to 4 weeks (by definition, all reach nadir by 4 weeks; 50% by week 1, 80% by week 2, 90% by week 3)
- Plateau phase: 2-4 weeks (or longer)
- Recovery phase: May take months to years
Outcomes:
- ~15%: Mild course, remain ambulatory, recover within weeks
- ~70%: Complete recovery within 12 months; 82% within 24 months
- ~20%: Residual motor weakness at 1 year
- ~5-20%: Fulminant quadriplegia requiring ventilation, often with poor recovery
- 2-5%: Mortality (respiratory failure, autonomic instability, infections)
- ~5%: Recurrence after recovery
Poor Prognostic Factors:
- Age >60 years (less relevant in pediatrics)
- Preceding diarrheal illness (C. jejuni)
- CMV infection
- Rapid progression to nadir (<7 days)
- Need for ventilatory support
- Low distal CMAP amplitudes (<20% of lower limit of normal) or inexcitable nerves
- Hyponatremia
- AMSAN subtype
GBS Disability Scale (for grading severity):
| Grade | Description |
|---|---|
| 0 | Normal |
| 1 | Minor symptoms, runs normally |
| 2 | Walks 5m unaided |
| 3 | Walks 5m with aid |
| 4 | Bedbound/chairbound |
| 5 | Requires ventilation |
| 6 | Death |
COMPLICATIONS
| Complication | Notes |
|---|---|
| Respiratory failure | 30% require mechanical ventilation |
| Autonomic dysfunction | Present in 65%; most dangerous in first 2-4 weeks; arrhythmias, BP swings, asystole |
| Aspiration pneumonia | From bulbar weakness |
| Deep vein thrombosis / PE | Immobility |
| Pressure ulcers | Immobility |
| Pain | Severe in 70%; often undertreated |
| Hyponatremia | SIADH; worsens prognosis |
| Post-GBS fatigue | Persistent in many patients |
| Residual weakness / deformity | Especially if AMSAN |
SPECIAL NOTE FOR PEDIATRICS
- Pain (leg pain, back pain) is a very prominent and early feature in children, often preceding weakness - can be misdiagnosed as "growing pains" or musculoskeletal pain
- Children frequently present with refusal to walk or unexplained abdominal pain as the first complaint
- Ataxia is often more prominent in children than in adults
- Autonomic dysfunction: urinary retention present in 10-15% early in the course
- 10-12% of children may require mechanical ventilation
- Recovery tends to be better in children than in adults
- AMAN subtype is more common in Asian children (especially after C. jejuni gastroenteritis)
SUMMARY BOX
| Feature | Classic GBS (AIDP) |
|---|---|
| Onset | 1-4 weeks after prodrome |
| Pattern | Symmetric ascending flaccid weakness |
| Reflexes | Absent/hypo-reflexic |
| Sensory | Mild distal; vibration sense ↓ |
| Cranial nerve | Bifacial palsy in 45-75% |
| Autonomic | 65% (BP, HR, bowel, bladder) |
| CSF | Albumin ↑↑, cells <10 (albuminocytological dissociation) |
| NCS | Conduction slowing, prolonged F-waves |
| Treatment | IVIG 2 g/kg OR Plasma exchange (both equally effective) |
| NOT used | Corticosteroids |
| Prognosis | 70% recover by 12 months; 2-5% mortality |
References:
- Bradley & Daroff's Neurology in Clinical Practice - GBS (Chapter 106)
- Goldman-Cecil Medicine International Edition - GBS Treatment
- Harriet Lane Handbook (23e), Johns Hopkins - Pediatric GBS
- Fishman's Pulmonary Diseases - Respiratory Management in GBS
- Braunwald's Heart Disease - Autonomic Complications
- WHO/Iran Journal of Child Neurology - Childhood GBS review
This is a shared conversation. Sign in to Orris to start your own chat.