Anti dyslipidemic drug
In detail
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Searching PubMed
dyslipidemia treatment guidelines lipid lowering therapy
Reading File
Antidyslipidemic Drugs - Detailed Overview
Background: Lipoproteins and Dyslipidemia
Dyslipidemia refers to abnormal levels of lipids or lipoproteins in the blood, including elevated LDL-cholesterol, elevated triglycerides, low HDL-cholesterol, or combinations. These abnormalities are major drivers of atherosclerotic cardiovascular disease (ASCVD). Drug therapy principally targets lowering LDL-cholesterol and/or non-HDL-cholesterol, as reduction of these is most strongly linked to cardiovascular event reduction.
"Lipid-modifying drugs that predominantly lower LDL-cholesterol and/or non-HDL-cholesterol levels are the first-line approach to reduce the risk of a first or recurrent cardiovascular event." - Goldman-Cecil Medicine, 2-Volume Set
Classification of Antidyslipidemic Drugs
| Class | Examples | Primary Effect |
|---|---|---|
| HMG-CoA reductase inhibitors (Statins) | Atorvastatin, Rosuvastatin, Simvastatin, Lovastatin | ↓↓↓ LDL-C |
| Cholesterol absorption inhibitors | Ezetimibe | ↓ LDL-C |
| PCSK9 monoclonal antibodies | Alirocumab, Evolocumab | ↓↓↓ LDL-C |
| PCSK9 siRNA | Inclisiran | ↓↓ LDL-C |
| Bile acid sequestrants | Cholestyramine, Colestipol, Colesevelam | ↓ LDL-C |
| Fibrates | Fenofibrate, Gemfibrozil | ↓↓ TG, ↑ HDL |
| Niacin (Nicotinic acid) | Extended-release niacin | ↓ TG, ↑ HDL, ↓ LDL |
| Omega-3 fatty acids | Icosapentaenoic acid (IPE), DHA/EPA | ↓↓ TG |
| ATP-citrate lyase inhibitor | Bempedoic acid | ↓ LDL-C |
| ANGPTL3 inhibitor | Evinacumab | ↓↓ LDL-C (homozygous FH) |
1. HMG-CoA Reductase Inhibitors (Statins)
Mechanism
Statins competitively inhibit HMG-CoA reductase, the rate-limiting enzyme in the mevalonate pathway for cholesterol biosynthesis in the liver. This reduces intracellular cholesterol, upregulates hepatic LDL receptors, and increases LDL clearance from plasma.
Drugs and Intensity Classification
| Intensity | Drugs | Expected LDL-C Reduction |
|---|---|---|
| High | Atorvastatin 40-80 mg, Rosuvastatin 20-40 mg | ≥50% |
| Moderate | Atorvastatin 10-20 mg, Rosuvastatin 5-10 mg, Simvastatin 20-40 mg, Pravastatin 40-80 mg, Lovastatin 40 mg | 30-50% |
| Low | Simvastatin 10 mg, Pravastatin 10-20 mg, Lovastatin 20 mg | <30% |
Pleiotropic Effects (Beyond Lipid Lowering)
- Anti-inflammatory effects (↓ CRP)
- Improved endothelial function
- Plaque stabilization
- Antithrombotic effects
Cardiovascular Outcomes Evidence
Statins reduce major cardiovascular events (MI, stroke, cardiovascular death) by ~25-35% per 1 mmol/L reduction in LDL-C. This is the most evidence-backed class in all of medicine, across primary and secondary prevention. They also reduce all-cause mortality.
Adverse Effects
- Myopathy - the most significant concern. Ranges from myalgia (muscle pain without CK elevation) to myositis (with elevated CK) to rhabdomyolysis (severe, rare - CK >10× ULN, myoglobinuria, renal failure risk). Risk factors: high dose, CYP3A4 inhibitors (for lovastatin/simvastatin/atorvastatin), hypothyroidism, renal impairment, elderly, drug interactions.
- Hepatotoxicity - transaminase elevation; clinical hepatitis is rare. Monitoring recommended if >5× ULN.
- New-onset diabetes - small but significant increased risk (~10%); benefit of ASCVD prevention overwhelmingly outweighs this risk.
- Nocebo effect - >75-80% of statin "intolerance" symptoms in blinded trials are not due to the statin.
Drug Interactions (Important)
- CYP3A4 inhibitors (azole antifungals, macrolides, HIV protease inhibitors, amiodarone) increase plasma levels of lovastatin, simvastatin, atorvastatin → increased myopathy risk
- Gemfibrozil inhibits glucuronidation of statin lactones → increased myopathy risk (avoid with cerivastatin; caution with all statins)
- Cyclosporine, danazol increase statin levels via OATP1B1 inhibition
- Goldman-Cecil Medicine, International Edition
2. Cholesterol Absorption Inhibitor - Ezetimibe
Mechanism
Ezetimibe inhibits NPC1L1 (Niemann-Pick C1-like 1) protein in the brush border of small intestinal enterocytes, blocking dietary and biliary cholesterol absorption. It does NOT affect triglyceride or fat-soluble vitamin absorption.
Efficacy
- Reduces LDL-C by ~18-20% as monotherapy
- Added to a statin: additional ~20-25% LDL-C reduction
- Has low triglyceride-lowering effect (~9%)
- No significant effect on HDL-C
Clinical Evidence
The IMPROVE-IT trial showed that adding ezetimibe to simvastatin in post-ACS patients further reduced cardiovascular events by ~6.4% vs placebo, confirming the "lower is better" hypothesis for LDL-C.
Dosing
10 mg once daily, orally. Can be taken at any time.
Adverse Effects
Generally well tolerated. Rare myopathy when combined with statins. Mild GI effects.
- Goldman-Cecil Medicine; Ezetimibe section
3. PCSK9 Inhibitors (Monoclonal Antibodies)
Mechanism
PCSK9 (Proprotein Convertase Subtilisin/Kexin type 9) is a serine protease that binds to LDL receptors on hepatocytes and targets them for lysosomal degradation. Anti-PCSK9 monoclonal antibodies (alirocumab, evolocumab) block this binding, allowing LDL receptors to recycle to the hepatocyte surface and clear more LDL from circulation.
- Genetic basis: Loss-of-function PCSK9 mutations → lower LDL-C, markedly reduced ASCVD risk. Gain-of-function mutations → familial hypercholesterolemia.
Drugs
| Drug | Dose | LDL-C Reduction |
|---|---|---|
| Alirocumab (Praluent) | 75 mg or 150 mg SC q2wk | 40-60% |
| Evolocumab (Repatha) | 140 mg SC q2wk or 420 mg SC q4wk | 55-65% |
Cardiovascular Outcomes
- FOURIER trial (Evolocumab): ~15% relative risk reduction in major ASCVD events
- ODYSSEY OUTCOMES (Alirocumab): Significant MACE reduction; all-cause mortality reduction in high-risk patients
Indications (2018 AHA/ACC guidelines)
Used in patients with:
- Established ASCVD with LDL-C ≥70 mg/dL despite maximally tolerated statin + ezetimibe
- Familial hypercholesterolemia with LDL-C ≥70 mg/dL (secondary prevention) or ≥100 mg/dL (primary prevention)
- Homozygous familial hypercholesterolemia
Adverse Effects
Injection site reactions (mild). Nasopharyngitis, flu-like symptoms. Generally excellent tolerability. Neurocognitive effects were investigated and found not clinically significant.
- Goldman-Cecil Medicine, International Edition
4. PCSK9 siRNA - Inclisiran
Mechanism
Inclisiran is a small interfering RNA (siRNA) that targets the hepatic mRNA encoding PCSK9, reducing PCSK9 synthesis in the liver (rather than blocking its action in the blood like the monoclonal antibodies).
Advantages
- Subcutaneous injection every 6 months (after initial loading doses at 0, 3 months)
- Reduces LDL-C by 40-50%
- Sustained effects over at least 18 months
- Long dosing interval improves adherence
- Goldman-Cecil Medicine; Inclisiran section
5. Bile Acid Sequestrants (Resins)
Mechanism
Bile acid sequestrants (cholestyramine, colestipol, colesevelam) are large, positively charged polymers that bind bile acids in the intestinal lumen, preventing their enterohepatic reabsorption. This forces the liver to convert more cholesterol into bile acids (via cholesterol 7-alpha hydroxylase), increasing LDL receptor expression and LDL uptake.
Efficacy
- Reduce LDL-C by 15-25% (dose-dependent)
- May slightly raise triglycerides - caution in hypertriglyceridemia
- Modestly raise HDL-C
Adverse Effects
- GI side effects: constipation, bloating, flatulence, nausea (major limitation)
- Drug interactions - bind concurrently administered drugs (warfarin, digoxin, thyroid hormones, thiazides, fat-soluble vitamins). Must administer other drugs 1 hour before or 4-6 hours after.
- Colesevelam has fewer GI side effects and is also FDA-approved for type 2 diabetes (lowers HbA1c ~0.5%)
Special Use
Safe in pregnancy (not systemically absorbed). First-line in children with familial hypercholesterolemia.
6. Fibrates (Fibric Acid Derivatives)
Mechanism
Fibrates activate PPAR-alpha (peroxisome proliferator-activated receptor alpha) in the liver and muscle, which:
- Increases lipoprotein lipase (LPL) synthesis → ↑ TG-rich lipoprotein catabolism
- Reduces apoC-III synthesis → ↓ inhibition of LPL
- Increases apoA-I and apoA-II synthesis → ↑ HDL-C
- Modestly reduces LDL-C (mainly in mixed dyslipidemia)
Drugs and Efficacy
| Drug | TG Reduction | HDL Increase | LDL Change |
|---|---|---|---|
| Fenofibrate 145 mg | 15-36% (mixed) | +5 to +15% | Modest ↓ |
| Gemfibrozil 600 mg BID | 46-62% (isolated hypertriglyceridemia) | +15 to +20% | Modest ↓ |
Cardiovascular Outcomes
Fibrates have failed to show mortality benefit in large trials when added to statins (ACCORD-LIPID). May benefit patients with combined TG ≥200 + HDL-C <40 mg/dL (subgroup analysis). Not first-line for ASCVD prevention.
Adverse Effects
- Myopathy (especially gemfibrozil + statin combination - pharmacokinetic interaction: avoid)
- Hepatotoxicity (transaminase elevation)
- Gallstones (↑ lithogenicity of bile)
- Renal: modest rise in serum creatinine (fenofibrate)
Drug Interactions
Gemfibrozil + statins = significant myopathy risk (gemfibrozil inhibits CYP2C8 and OATP transporters responsible for statin glucuronidation). Fenofibrate is much safer with statins.
Warfarin - fibrates potentiate anticoagulant effect; close INR monitoring required.
7. Niacin (Nicotinic Acid)
Mechanism
At pharmacologic doses, niacin acts via the GPR109A receptor (HCAR2) on adipocytes to inhibit hormone-sensitive lipase → ↓ free fatty acid (FFA) release from adipose tissue → ↓ hepatic VLDL synthesis → ↓ TG, ↓ LDL-C. Also directly inhibits hepatic VLDL secretion.
Efficacy
- ↓ TG by 20-50%
- ↑ HDL-C by 15-35% (most effective agent for raising HDL)
- ↓ LDL-C by 15-25%
- Also reduces Lp(a) by ~25-30%
Clinical Outcomes
Despite favorable lipid changes, the AIM-HIGH and HPS2-THRIVE trials failed to show added ASCVD benefit when niacin was added to statin therapy (with adequate LDL-C lowering). As a result, the use of niacin has sharply declined and it is not recommended as first-line add-on therapy.
Adverse Effects
- Flushing - prostaglandin-mediated, most common side effect; can be reduced by taking aspirin 30 minutes before, using extended-release formulation, taking at bedtime, avoiding hot beverages/alcohol.
- Hepatotoxicity - especially with immediate-release niacin at high doses
- Hyperglycemia - worsens insulin resistance
- Hyperuricemia / gout exacerbation
- GI: nausea, diarrhea
- Acanthosis nigricans (skin darkening)
8. Omega-3 Fatty Acids
Mechanism
At high pharmacologic doses (4 g/day), reduce VLDL synthesis and secretion from the liver, enhance TG clearance via LPL. Two distinct preparations exist with different effects:
| Agent | Composition | TG Reduction | LDL-C Effect |
|---|---|---|---|
| EPA + DHA (Lovaza, Vascepa combo) | EPA + DHA | 26-52% (isolated hypertriglyceridemia) | May raise LDL-C slightly |
| Icosapentaenoic acid (IPE/EPA only) - Vascepa | Pure EPA | 19-44% | No LDL-C rise |
Cardiovascular Evidence
The REDUCE-IT trial (Vascepa/IPE 4 g/day) showed a remarkable 25% relative risk reduction in major cardiovascular events in patients with elevated TG (≥150 mg/dL) on statin therapy, at high CV risk. This is one of the most significant recent cardiovascular outcomes results.
Adverse Effects
Generally well tolerated. GI side effects (fishy aftertaste, GI discomfort), mild bleeding time prolongation. Atrial fibrillation risk (slight increase with high-dose omega-3 formulations).
9. Bempedoic Acid
Mechanism
Bempedoic acid is an oral prodrug activated by very long-chain acyl-CoA synthetase 1 (ACSVL1) specifically in the liver (not in skeletal muscle, unlike statins). It inhibits ATP-citrate lyase (ACL), an enzyme upstream of HMG-CoA reductase in cholesterol synthesis.
- Because activation occurs only in hepatocytes, skeletal muscle is spared → lower risk of myopathy than statins.
Efficacy
- Reduces LDL-C by ~18-25% vs placebo
- Combined with ezetimibe (Nexlizet): reduces LDL-C by ~38%
- ~12% reduction in subsequent cardiovascular events
Key Role
Particularly useful in statin-intolerant patients who need additional LDL-C lowering. The CLEAR Outcomes trial demonstrated cardiovascular event reduction.
Adverse Effects
Gout/hyperuricemia (blocks renal tubular URAT1, increases uric acid), myalgia (rare, less than statins), tendon rupture (black box warning - rare).
- Goldman-Cecil Medicine, International Edition
10. Evinacumab (ANGPTL3 Inhibitor)
Mechanism
Evinacumab is a fully humanized monoclonal antibody that inhibits angiopoietin-like protein 3 (ANGPTL3), which normally inhibits lipoprotein lipase and endothelial lipase. Blocking ANGPTL3 allows greater TG hydrolysis and lipoprotein clearance.
Indication
Primarily for homozygous familial hypercholesterolemia (HoFH) - reduces LDL-C by >50% even in patients with no functional LDL receptors (works via an LDL receptor-independent pathway).
Triglyceride-Lowering Drug Efficacy Comparison
From Goldman-Cecil Medicine (E-Table 190-1):
| Drug | TG Reduction |
|---|---|
| Gemfibrozil 600 mg BID (isolated hypertriglyceridemia) | 46-62% |
| Marine omega-3 FA (dose-dependent, isolated hypertriglyceridemia) | 26-52% |
| Niacin ER 2g (mixed dyslipidemia) | 5-38% |
| Fenofibrate 145 mg | 15-36% |
| Marine omega-3 FA (mixed dyslipidemia) | 19-44% |
| Ezetimibe 10 mg | ~9% |
Approach to Statin Intolerance
From Goldman-Cecil Medicine (Table 190-5):
Mild-moderate symptoms:
- Stop statin
- Wait until symptoms resolve
- Rechallenge with lower dose of same statin, or alternative statin
- Titrate up as tolerated
Severe muscle pain/weakness (rhabdomyolysis suspected):
- Stop statin
- Check CK, creatinine/GFR, urine myoglobin
- Admit if CK >10× ULN with elevated creatinine or myoglobinuria
- Evaluate predisposing factors
Blinded rechallenge studies show >75-80% of patients reporting "statin intolerance" can tolerate atorvastatin 20 mg as blinded therapy - the "nocebo effect" is a major contributor.
Treatment Algorithm (2018 AHA/ACC Guidelines)
Step 1: Lifestyle modification (diet, exercise)
↓
Step 2: Statin (first-line; high-intensity preferred for high/very high risk)
↓
Step 3: If LDL-C remains ≥70 mg/dL on maximally tolerated statin:
Add Ezetimibe
↓
Step 4: If still inadequate:
Add PCSK9 inhibitor (alirocumab or evolocumab)
OR Inclisiran (every 6 months dosing)
OR Bempedoic acid (especially if statin-intolerant)
↓
Hypertriglyceridemia (TG ≥500 mg/dL):
Fibrate or Omega-3 FA to prevent pancreatitis
↓
Icosapentaenoic acid (Vascepa):
For residual TG elevation (≥150 mg/dL) on statin at high CV risk
Special Populations
| Condition | Preferred Agent(s) | Avoid/Caution |
|---|---|---|
| Pregnancy | Bile acid sequestrants | All other agents (statins contraindicated) |
| Familial hypercholesterolemia (heterozygous) | High-intensity statin + ezetimibe ± PCSK9 inhibitor | - |
| Homozygous FH | Statin + ezetimibe + PCSK9 inhibitor + evinacumab | - |
| Statin-intolerant | Bempedoic acid ± ezetimibe, PCSK9 inhibitor | Gemfibrozil + statin |
| Isolated hypertriglyceridemia (TG >500) | Fibrate or omega-3 FA | - |
| CKD | Atorvastatin, rosuvastatin (dose adjust); avoid gemfibrozil | - |
| Liver disease | Caution with all; bile acid sequestrants relatively safer | - |
Recent Evidence (2025-2026)
- A 2025 network meta-analysis in J Cardiovasc Pharmacol (PMID 40358978) compared the efficacy of non-statin lipid-lowering therapies, showing the comparative superiority of PCSK9 inhibitors over ezetimibe and other agents for LDL-C reduction.
- A 2025 meta-analysis in Atherosclerosis (PMID 40618457) examined effects of various lipid-lowering therapies on Lp(a), finding that PCSK9 inhibitors significantly reduce Lp(a) by 20-30%, a benefit not seen with statins.
- PCSK9 inhibitors now have evidence for peripheral arterial disease reduction as well (2026 systematic review).
Sources: Goldman-Cecil Medicine International Edition, 2-Volume Set; Katzung's Basic and Clinical Pharmacology, 16th Edition; recent PubMed meta-analyses (PMIDs 40358978, 40618457, 41895598)
Prostaglandin from lip Pinchot
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Loading Image
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Lippincot pharmacology
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Reading File
Finding Sources
I now have all the content needed. Here is the complete, detailed Lippincott Pharmacology chapter on Prostaglandins:
Prostaglandins - Lippincott Illustrated Reviews: Pharmacology
Overview
Prostaglandins (PGs) are unsaturated fatty acid derivatives containing 20 carbons that include a cyclic ring structure. They are sometimes referred to as eicosanoids (from "eicosa" = 20 carbons). NSAIDs act through inhibition of prostaglandin synthesis, so understanding prostaglandins is essential to understanding NSAID pharmacology.
I. Role of Prostaglandins as Local Mediators
- Prostaglandins are produced in minute quantities by virtually all tissues
- They act locally on the tissues in which they are synthesized (paracrine/autocrine)
- Rapidly metabolized to inactive products at their sites of action
- Therefore, prostaglandins do not circulate in the blood in significant concentrations
- Thromboxanes and leukotrienes are related compounds synthesized from the same precursors
II. Synthesis of Prostaglandins
Step 1 - Release of Arachidonic Acid
Arachidonic acid is the primary precursor. It is normally stored as a component of cell membrane phospholipids. Free arachidonic acid is released by the action of phospholipase A2, a process triggered by hormones and other stimuli (including cell injury, inflammation).
Glucocorticoids inhibit phospholipase A2 (via lipocortin/annexin-1), reducing arachidonic acid availability - one mechanism of their powerful anti-inflammatory effect.
Step 2 - Two Major Pathways
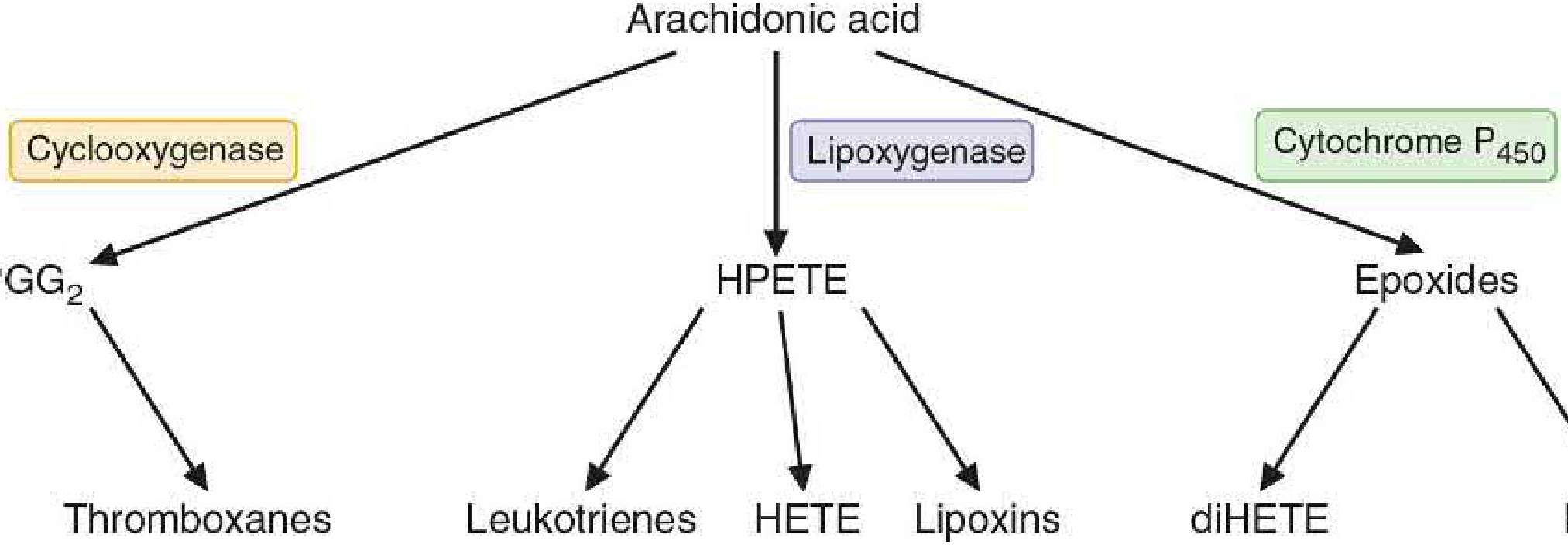
A. Cyclooxygenase (COX) Pathway - Eicosanoids with Ring Structures
This pathway produces prostaglandins, thromboxanes, and prostacyclin.
COX-1 vs COX-2
| Feature | COX-1 | COX-2 |
|---|---|---|
| Expression | Constitutive (always present) | Inducible (upregulated in inflammation) |
| Location | Virtually all tissues; only form in mature platelets | Brain, kidney, bone constitutively; induced elsewhere |
| Function | Gastric cytoprotection, vascular homeostasis, platelet aggregation, renal and reproductive function | Elevated prostanoid production at sites of inflammation and chronic disease |
| Binding site | Smaller, less flexible | Larger and more flexible substrate channel |
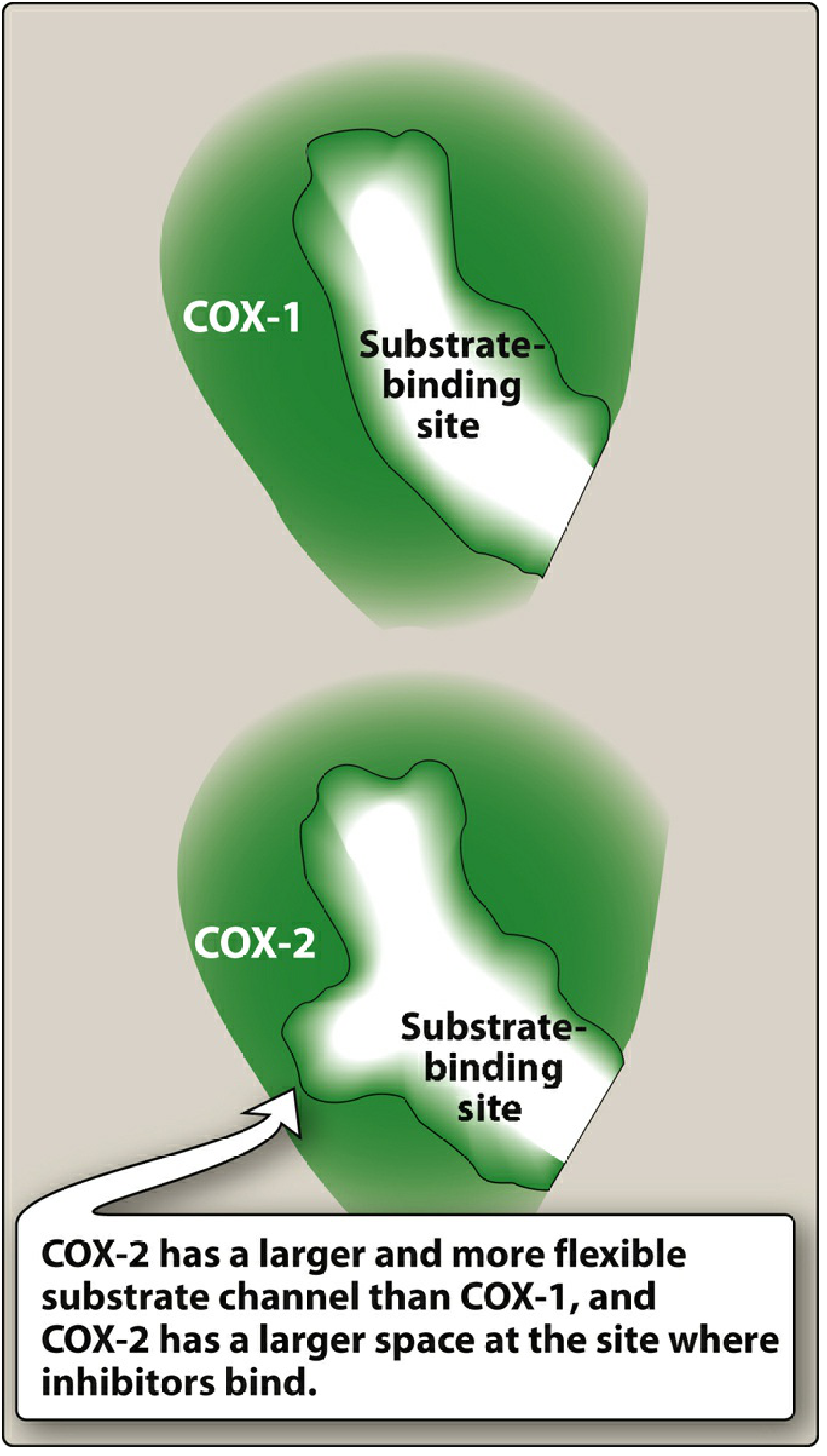
COX-2 has a larger and more flexible substrate channel than COX-1, with a larger space at the inhibitor binding site - this difference allowed the development of selective COX-2 inhibitors (celecoxib).
COX-2 expression is induced by inflammatory mediators (TNF-α, IL-1) and inhibited by glucocorticoids.
Biosynthetic Steps (COX Pathway)
- Cyclooxygenase acts on arachidonic acid → forms the 5-membered ring + adds 4 oxygen atoms → unstable endoperoxide PGG₂
- Peroxidase reduces hydroperoxy group at C-15 → PGH₂ (using glutathione as reducing agent)
- PGH₂ is the branch point - downstream products depend on tissue-specific enzymes:
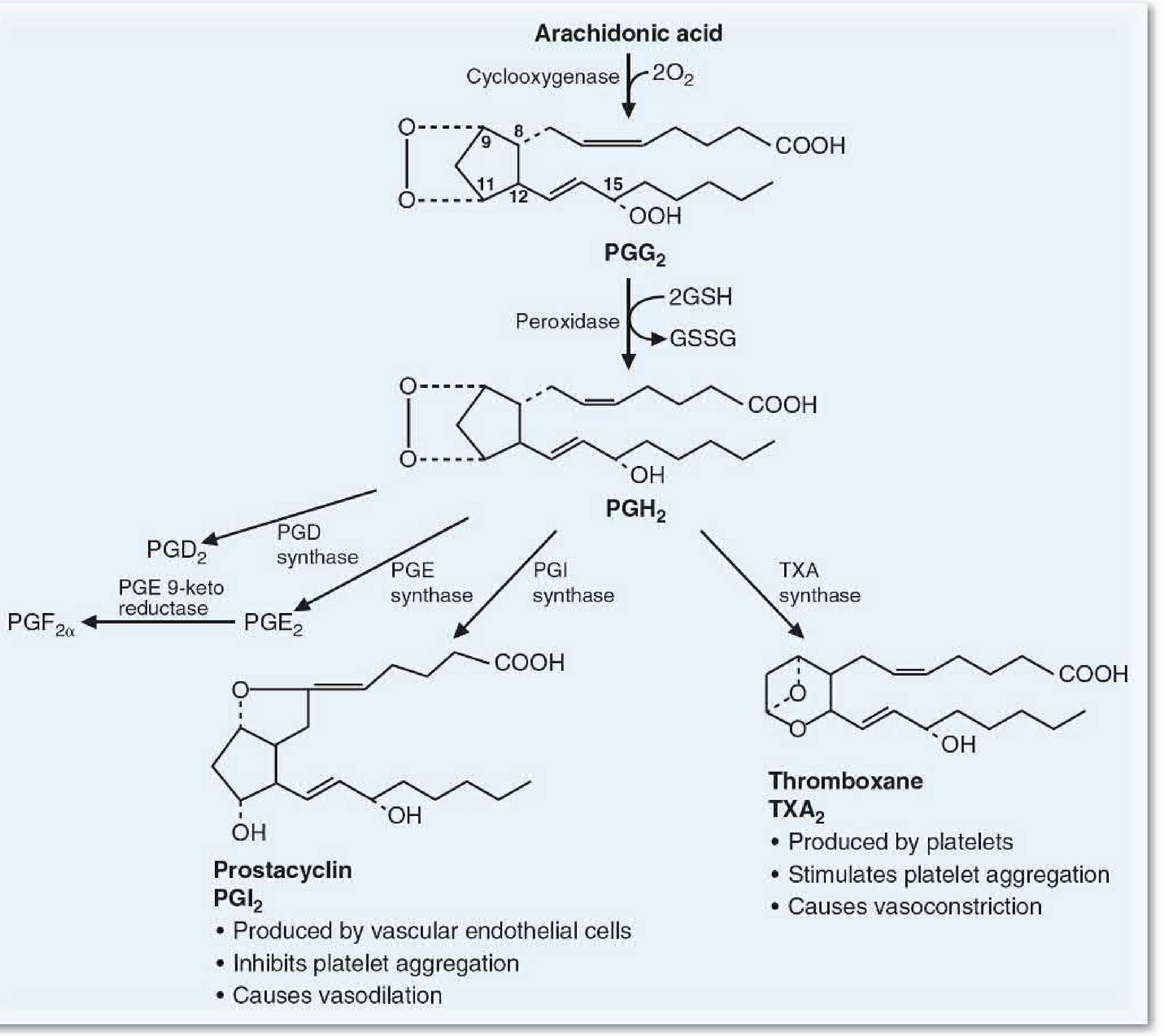
| Enzyme | Product | Source Tissue |
|---|---|---|
| PGE synthase | PGE₂ | Widely distributed |
| PGD synthase | PGD₂ | Mast cells, brain |
| PGE 9-ketoreductase / endoperoxide reductase | PGF₂α | Uterus, lungs |
| PGI synthase | PGI₂ (Prostacyclin) | Vascular endothelial cells |
| TXA synthase | TXA₂ (Thromboxane A₂) | Platelets |
B. Lipoxygenase (LOX) Pathway
Lipoxygenases act on arachidonic acid to form leukotrienes (via HPETE intermediate). These are important mediators of asthma and allergic reactions.
- Zileuton inhibits 5-lipoxygenase (blocks leukotriene synthesis)
- Zafirlukast, Montelukast - leukotriene receptor antagonists (used in asthma)
III. Actions of Prostaglandins
Actions are mediated by binding to G-protein coupled receptors (GPCRs) on cell membranes. The specific effect depends on which tissue and which receptor subtype is present.
Key Functional Dichotomy: TXA₂ vs PGI₂
| TXA₂ (Thromboxane A₂) | PGI₂ (Prostacyclin) | |
|---|---|---|
| Produced by | Platelets | Vascular endothelial cells |
| Effect on platelets | Stimulates aggregation | Inhibits aggregation |
| Vascular effect | Vasoconstriction | Vasodilation |
| cAMP effect | Decreases | Increases |
The net effect on platelets and blood vessels depends on the balance between TXA₂ and PGI₂.
Summary of Prostaglandin Actions by Type
| Prostaglandin | Major Actions |
|---|---|
| PGE₂ | Vasodilation, ↓ platelet aggregation, ↑ uterine contractions, fever induction (via hypothalamus), pain sensitization (sensitizes nociceptors to bradykinin/histamine), ↓ gastric acid, ↑ mucus |
| PGI₂ (Prostacyclin) | Vasodilation, ↓ platelet aggregation, bronchodilation |
| PGD₂ | Vasodilation, ↓ platelet aggregation, bronchoconstriction (in asthma) |
| PGF₂α | Vasoconstriction, bronchoconstriction, smooth muscle contraction, luteolysis |
| TXA₂ | Vasoconstriction, ↑ platelet aggregation, bronchoconstriction |
Role in Pain, Fever, and Inflammation
- Pain: PGE₂ sensitizes peripheral nerve endings to bradykinin, histamine, and other inflammatory mediators → lowers threshold for nociception
- Fever: Endogenous pyrogens (cytokines) → ↑ PGE₂ in hypothalamus → elevates thermoregulatory set-point → fever. NSAIDs reverse this by blocking PGE₂.
- Inflammation: Prostaglandins promote vasodilation, vascular permeability, and leukocyte recruitment
IV. Therapeutic Uses of Prostaglandins
Prostaglandins control acid secretion and mucus production (GI), uterine contractions, and renal blood flow. They are chemical mediators in allergic and inflammatory processes.
V. Prostaglandin Drug Classes
A. PGE₁ Analogs
1. Alprostadil (Caverject, Edex, MUSE, Prostin VR)
- Naturally produced in seminal vesicles, cavernous tissue, placenta, and ductus arteriosus
- PGE₁ maintains patency of the ductus arteriosus during fetal life
- Clinical uses:
- Neonates with congenital heart defects - IV infusion keeps ductus arteriosus open while awaiting surgical correction
- Erectile dysfunction - intracavernosal injection or intraurethral suppository
2. Lubiprostone (Amitiza)
- PGE₁ derivative
- Stimulates chloride channels (ClC-2) in intestinal epithelial luminal cells → ↑ intestinal fluid secretion
- Uses: Chronic idiopathic constipation, opioid-induced constipation, IBS with constipation
- Adverse effects: Nausea (most common - reduced if taken with food), diarrhea
3. Misoprostol (Cytotec)
- Synthetic PGE₁ analog (acid-stable, orally bioavailable - unlike natural PGE₁)
- Mechanism: Binds prostaglandin receptors on gastric parietal cells → ↓ gastric acid secretion + ↑ mucus and bicarbonate production
- Uses:
- Prevention of NSAID-induced gastric ulcers (combined with NSAIDs, including diclofenac-misoprostol fixed combination)
- Off-label obstetric use: Cervical ripening and labor induction (stimulates uterine contractions via uterine prostaglandin receptors)
- Contraindicated in pregnancy (can induce abortion)
- Adverse effects: Diarrhea, abdominal cramps
B. PGE₂ Analogs
Dinoprostone (Cervidil, Prepidil)
- Synthetic PGE₂ analog
- Uses: Cervical ripening and labor induction
- Promotes uterine smooth muscle contractions
C. PGF₂α Analogs (Prostaglandin Analogs for Glaucoma)
These agents lower intraocular pressure (IOP) by increasing uveoscleral outflow of aqueous humor.
| Drug | Brand | Key Notes |
|---|---|---|
| Latanoprost | Xalatan | Once-daily eye drops; first in class |
| Bimatoprost | Lumigan, Latisse | Also approved for eyelash hypotrichosis (Latisse) |
| Tafluprost | Zioptan | Preservative-free formulation |
| Travoprost | Travatan Z | Preservative-free option |
Class adverse effects:
- Iris pigmentation (irreversible darkening of iris color - especially in hazel/green eyes)
- Eyelash changes (increased length, thickness, darkening)
- Conjunctival hyperemia
- Local irritation
D. PGI₂ (Prostacyclin) Analogs
Used for pulmonary arterial hypertension (PAH) - mimic PGI₂'s vasodilatory effects on pulmonary vasculature.
| Drug | Route | Notes |
|---|---|---|
| Epoprostenol (Flolan, Veletri) | IV (continuous infusion) | Very short half-life; requires central line |
| Iloprost (Ventavis) | Inhaled | Repeated inhalation sessions/day |
| Treprostinil (Tyvaso, Remodulin) | Inhaled or SC/IV | Longer half-life than epoprostenol |
Mechanism: Mimic prostacyclin effects in endothelial cells → significant reduction in pulmonary arterial resistance → ↑ cardiac index and oxygen delivery.
VI. Pharmacological Inhibition of Prostaglandin Synthesis
NSAIDs - Mechanism Summary
NSAIDs inhibit cyclooxygenase (COX-1 and/or COX-2), blocking the conversion of arachidonic acid to PGG₂/PGH₂ and downstream prostaglandins.
| Drug Class | COX Selectivity | Key Examples |
|---|---|---|
| Non-selective NSAIDs | COX-1 + COX-2 | Ibuprofen, Naproxen, Indomethacin, Diclofenac |
| Aspirin | COX-1 >> COX-2 (irreversible) | Irreversible acetylation of COX active site serine residue |
| Selective COX-2 inhibitors | COX-2 selective | Celecoxib |
Aspirin - Special Mechanism
Aspirin irreversibly acetylates the active site serine residue of COX, permanently inactivating the enzyme. Since platelets have no nucleus and cannot synthesize new COX, aspirin's antiplatelet effect (via TXA₂ blockade) lasts the entire platelet lifespan (7-10 days).
Clinical Consequences of COX Inhibition
| Effect | Mechanism via Prostaglandins |
|---|---|
| Analgesia | ↓ PGE₂ → reduced sensitization of nociceptors to bradykinin/histamine |
| Antipyresis | ↓ PGE₂ in hypothalamus → reset thermoregulatory set-point |
| Anti-inflammatory | ↓ vasodilation, vascular permeability, leukocyte recruitment |
| Antiplatelet (aspirin) | ↓ TXA₂ in platelets → reduced platelet aggregation |
| GI ulceration (side effect) | ↓ COX-1 → ↓ gastroprotective PGE₂ → ↓ mucus/bicarbonate, ↑ acid |
| Renal effects | ↓ renal prostaglandins → ↓ renal blood flow (especially in volume-depleted patients) |
| Cardiovascular risk (COX-2 selective) | ↓ endothelial PGI₂ without affecting platelet TXA₂ → prothrombotic state |
Summary Diagram of Clinical Applications
PROSTAGLANDIN ANALOGS:
├── PGE₁ (Alprostadil) ──→ Patent ductus arteriosus (neonates), Erectile dysfunction
├── PGE₁ (Lubiprostone) ──→ Constipation, IBS
├── PGE₁ (Misoprostol) ──→ Gastric ulcer prevention (anti-NSAID), Labor induction
├── PGE₂ (Dinoprostone) ──→ Cervical ripening, Labor induction
├── PGF₂α (Latanoprost, Bimatoprost, etc.) ──→ Glaucoma (↓ IOP)
└── PGI₂ (Epoprostenol, Iloprost, Treprostinil) ──→ Pulmonary arterial hypertension
PROSTAGLANDIN INHIBITORS (NSAIDs):
├── Non-selective (Ibuprofen, Naproxen) ──→ Anti-inflammatory, Analgesia, Antipyresis
├── Aspirin (irreversible) ──→ Antiplatelet, Analgesia, Antipyresis
└── COX-2 selective (Celecoxib) ──→ Anti-inflammatory with ↓ GI side effects
Source: Lippincott Illustrated Reviews: Pharmacology, 9781975170561
NSAID
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
Loading Image
Reading File
Loading Image
Loading Image
I now have everything needed for a comprehensive response. Here is the complete NSAID chapter from Lippincott Illustrated Reviews: Pharmacology:
NSAIDs (Non-Steroidal Anti-Inflammatory Drugs)
Lippincott Illustrated Reviews: Pharmacology
I. Overview & Classification
NSAIDs are a group of chemically dissimilar agents that share antipyretic, analgesic, and anti-inflammatory activity. They act primarily by inhibiting cyclooxygenase (COX) enzymes, blocking the first step in prostanoid biosynthesis.
Chemical Classes of NSAIDs
| Class | Drugs |
|---|---|
| Salicylates | Aspirin, Diflunisal, Salsalate |
| Propionic acids | Ibuprofen, Naproxen, Ketoprofen, Fenoprofen, Flurbiprofen, Oxaprozin |
| Acetic acids | Diclofenac, Indomethacin, Ketorolac, Etodolac, Sulindac, Nabumetone, Tolmetin |
| Enolic acids (Oxicams) | Meloxicam, Piroxicam |
| Fenamates | Mefenamic acid, Meclofenamate |
| Selective COX-2 inhibitor | Celecoxib |
Key principle: COX-2 inhibition → anti-inflammatory and analgesic actions. COX-1 inhibition → prevention of cardiovascular events but also most adverse effects (GI, renal, platelet).
II. COX-1 vs COX-2 Selectivity
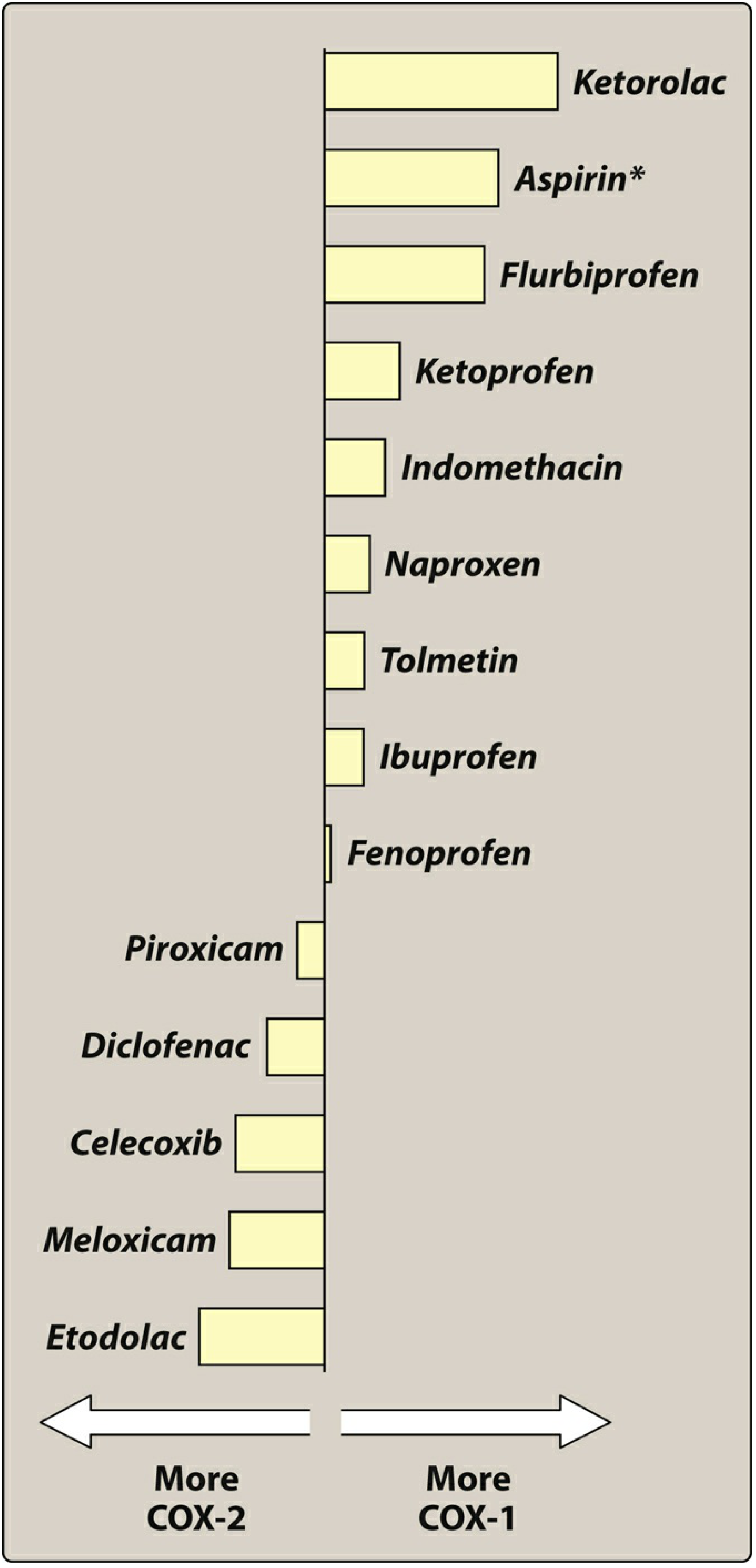
Most COX-1 selective (highest GI risk): Ketorolac > Aspirin > Flurbiprofen > Ketoprofen > Indomethacin > Naproxen > Ibuprofen
Most COX-2 selective (lowest GI risk): Etodolac > Meloxicam > Celecoxib > Diclofenac > Piroxicam
III. Mechanism of Action
All NSAIDs inhibit cyclooxygenase, blocking the conversion of arachidonic acid → PGG₂ → PGH₂, reducing synthesis of prostaglandins, thromboxanes, and prostacyclin.
Three Therapeutic Actions:
- Anti-inflammatory - ↓ PG-mediated vasodilation, vascular permeability, leukocyte recruitment
- Analgesic - ↓ PGE₂, which normally sensitizes nociceptors to bradykinin and histamine
- Antipyretic - ↓ PGE₂ synthesis in hypothalamus → reset of thermoregulatory set-point → ↑ heat dissipation (vasodilation + sweating); no effect on normal body temperature
IV. Aspirin - The Prototype
Unique Mechanism: Irreversible COX Inhibition
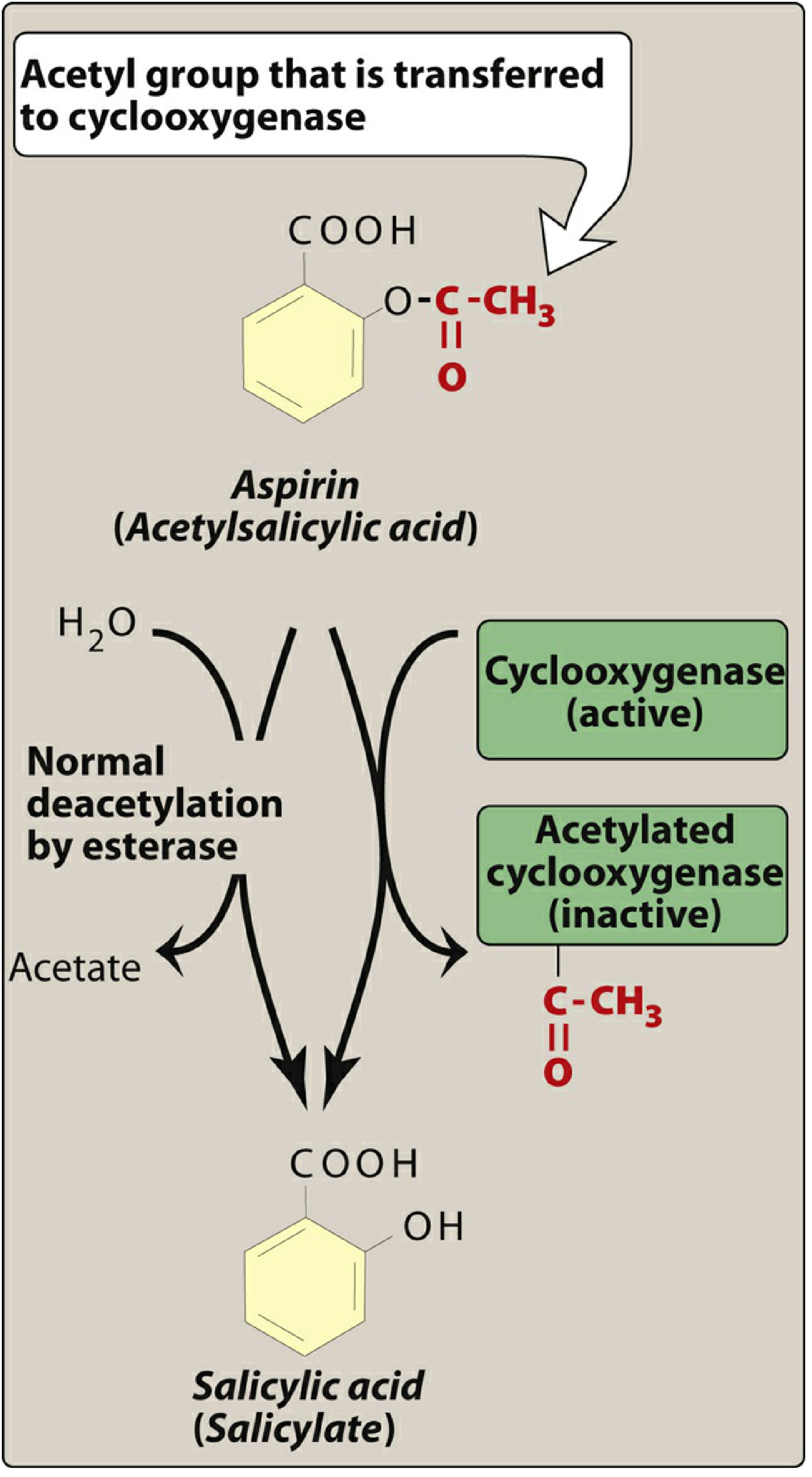
Aspirin is a weak organic acid that irreversibly acetylates a serine residue in the active site of COX-1 (and to a lesser extent COX-2), permanently inactivating it. All other NSAIDs are reversible inhibitors.
After acetylation, aspirin is hydrolyzed to salicylate by plasma esterases.
Aspirin Dose-Dependent Effects
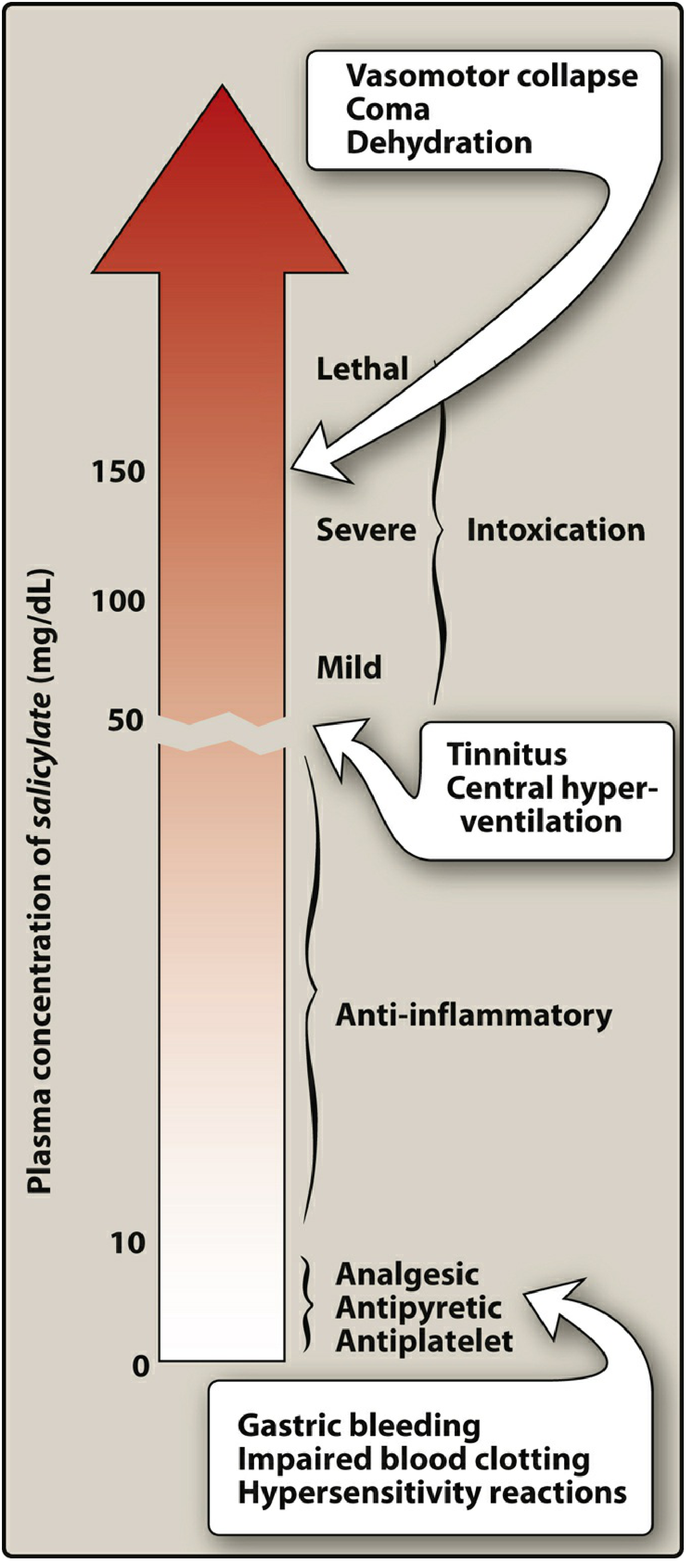
| Plasma Level | Effect |
|---|---|
| Low (~10 mg/dL) | Antiplatelet, Analgesic, Antipyretic |
| Moderate (~20-30 mg/dL) | Anti-inflammatory |
| ~50 mg/dL | Tinnitus, central hyperventilation (salicylism) |
| >100 mg/dL | Severe intoxication |
| >150 mg/dL | Lethal - vasomotor collapse, coma, dehydration |
V. Therapeutic Uses of NSAIDs
A. Anti-inflammatory and Analgesic Uses
- Osteoarthritis, rheumatoid arthritis (RA), gout
- Headache, arthralgia, myalgia, dysmenorrhea
- Combinations with opioids for cancer pain (opioid-sparing effect)
- Salicylates: analgesic at lower doses; anti-inflammatory only at higher doses (12-20 aspirin tablets/day)
B. Antipyretic Uses
- Aspirin, ibuprofen, naproxen for fever
- ⚠ Aspirin CONTRAINDICATED in patients < 19 years with viral infections (varicella, influenza) → risk of Reye syndrome (fulminating hepatitis + cerebral edema, often fatal)
- Acetaminophen is the preferred antipyretic for children
C. Cardiovascular Applications (Aspirin)
- Aspirin irreversibly inhibits COX-1-mediated TXA₂ production in platelets
- Since platelets are anucleate (cannot synthesize new COX), the antiplatelet effect lasts the entire platelet lifespan (3-7 days)
- Low-dose aspirin (81-325 mg/day): reduces risk of MI, stroke, TIA
- Used in both primary and secondary cardiovascular prevention
- ⚠ Concomitant NSAID use may prevent aspirin from binding COX → patients should take aspirin at least 30 minutes before the NSAID
D. External/Topical Uses
- Salicylic acid (topical): acne, corns, calluses, warts
- Methyl salicylate ("oil of wintergreen"): counterirritant in arthritis creams, sports rubs
- Diclofenac gel/solution: topical treatment of osteoarthritis (knees/hands)
- Ketorolac eye drops: seasonal allergic conjunctivitis, post-ocular surgery pain/inflammation
VI. Pharmacokinetics
Aspirin
- Rapidly deacetylated by esterases → salicylate (active)
- Absorbed mainly from the upper small intestine (unionized form)
- Crosses the blood-brain barrier and placenta
- Hepatic conjugation → water-soluble metabolites → renal excretion
- At analgesic doses (<4 g/day): first-order kinetics, half-life ~3.5 hours
- At anti-inflammatory doses (>4 g/day): hepatic pathway saturates → zero-order kinetics, half-life up to 12+ hours
Low-dose aspirin decreases uric acid excretion → avoid in gout or in patients taking probenecid.
Other NSAIDs
- Well absorbed orally
- Highly protein-bound in plasma
- Metabolized by the liver (mostly to inactive metabolites)
- Exceptions with active metabolites: nabumetone, sulindac
- Excreted primarily via urine
VII. Adverse Effects
A. Gastrointestinal (Most Common)
Mechanism: COX-1 inhibition → ↓ PGI₂, PGE₂, PGF₂α → ↓ gastric mucus and bicarbonate → ↑ gastric acid → dyspepsia, gastritis, peptic ulceration, GI bleeding
- Take NSAIDs with food or fluids to reduce GI upset
- High-risk patients: add proton pump inhibitor (PPI) or misoprostol (PGE₁ analog - restores gastric protection)
B. Antiplatelet Effect / Bleeding
- All NSAIDs (not just aspirin) can prolong bleeding time
- Aspirin's antiplatelet effect: irreversible, lasts 3-7 days → withhold aspirin ≥1 week before surgery
- NSAIDs + anticoagulants: increased bleeding risk
C. Renal Effects
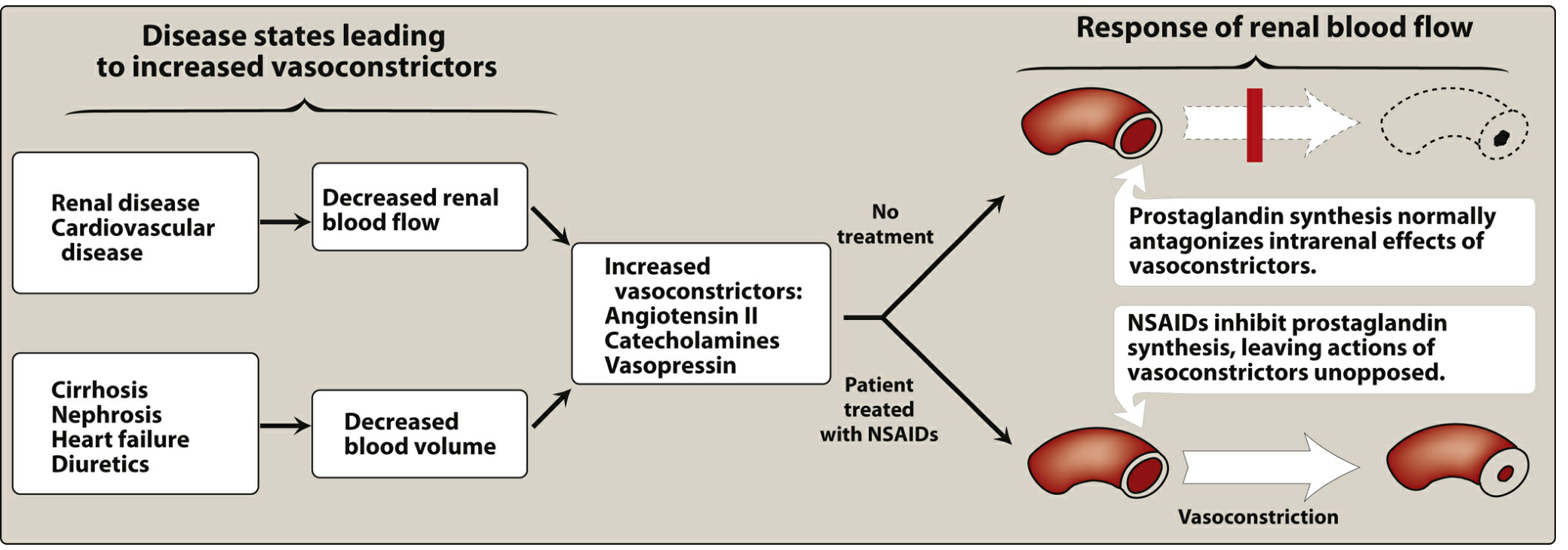
- ↓ PGE₂ and PGI₂ → ↓ renal blood flow → sodium and water retention, edema
- Can worsen heart failure, hypertension
- Blunts effects of antihypertensive medications
- May precipitate acute kidney injury in susceptible patients (renal disease, HF, volume depletion)
D. Cardiovascular Effects
- Aspirin (high COX-1 selectivity) → ↓ TXA₂ → cardioprotective
- COX-2 selective agents → ↓ endothelial PGI₂ WITHOUT reducing platelet TXA₂ → prothrombotic imbalance → ↑ risk of MI and stroke
- All NSAIDs except aspirin carry a Black Box Warning for increased cardiovascular risk
- In patients with cardiovascular disease who need an NSAID: naproxen is considered the least harmful
- NSAIDs other than aspirin: discouraged in established cardiovascular disease
E. Respiratory / Hypersensitivity
- NSAIDs block COX but NOT lipoxygenase → arachidonic acid is shunted into the leukotriene pathway → bronchoconstriction
- Aspirin-exacerbated respiratory disease (AERD) / Samter's triad: asthma + nasal polyps + NSAID sensitivity
- NSAIDs → ↑ leukotrienes → bronchospasm, rhinorrhea, urticaria
- Treatment: leukotriene receptor antagonists (zafirlukast, montelukast)
F. Salicylate Toxicity
Salicylism (mild toxicity): Nausea, vomiting, tinnitus, marked hyperventilation, headache, mental confusion, dizziness
Severe salicylate intoxication:
- Restlessness, delirium, hallucinations, convulsions, coma
- Mixed respiratory and metabolic acidosis
- Death from respiratory failure
- Children particularly susceptible (as little as 10 g can be fatal)
G. Pregnancy
- NSAIDs should only be used if benefits outweigh risks
- Third trimester: generally AVOID - risk of premature closure of the ductus arteriosus
- Acetaminophen preferred for analgesia/antipyresis in pregnancy
VIII. Drug Interactions
| Interacting Drug | Effect | Mechanism |
|---|---|---|
| Anticoagulants (warfarin) | ↑ Bleeding risk | Antiplatelet effect + GI irritation |
| Corticosteroids | ↑ GI ulceration risk | Additive GI damage |
| Methotrexate | ↑ Methotrexate toxicity | NSAIDs ↓ renal tubular secretion of MTX |
| ACE inhibitors / ARBs | ↓ Antihypertensive effect + ↑ AKI risk | ↓ Renal prostaglandins |
| Diuretics | ↓ Diuretic effect, ↑ AKI risk | ↓ Renal prostaglandins |
| Lithium | ↑ Lithium levels/toxicity | ↓ Renal lithium excretion |
| Probenecid (uricosuric) | Antagonism | Aspirin ↓ uric acid excretion at low doses |
| Other NSAIDs + aspirin | ↓ Aspirin's antiplatelet effect | NSAIDs compete for COX-1 binding |
IX. Selective COX-2 Inhibitor: Celecoxib
Mechanism
Celecoxib selectively inhibits COX-2 (reversible). It exploits the larger, more flexible substrate channel of COX-2 compared to COX-1.
Therapeutic Uses
- Osteoarthritis, RA, acute pain (similar analgesic efficacy to other NSAIDs)
Pharmacokinetics
- Well absorbed orally
- Metabolized by CYP2C9 in the liver
- Half-life ~11 hours (once or twice daily dosing)
- Reduce dose in moderate hepatic impairment; avoid in severe hepatic or renal disease
Advantages over Non-selective NSAIDs
- Less GI bleeding and dyspepsia (spares COX-1-mediated gastric protection)
- However, this benefit is lost when aspirin is co-administered
Disadvantages
- ↑ Cardiovascular risk (↓ endothelial PGI₂ without affecting platelet TXA₂)
- All NSAIDs (including celecoxib) carry a cardiovascular Black Box Warning
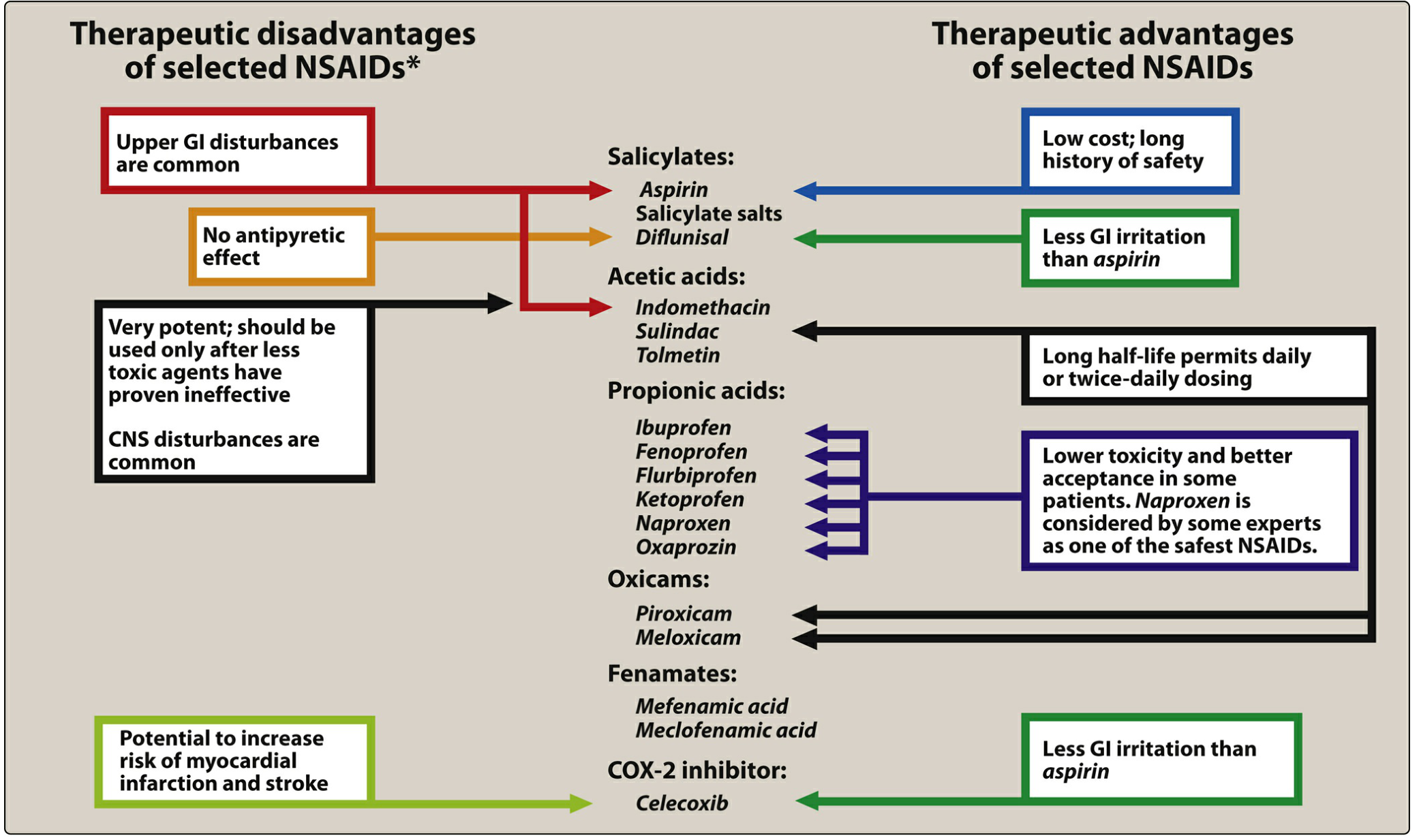
X. Summary of NSAID Advantages and Disadvantages
XI. Acetaminophen (Paracetamol) - For Comparison
Acetaminophen is NOT an NSAID but is frequently compared to them.
Mechanism
- Inhibits prostaglandin synthesis in the CNS → antipyretic + analgesic
- Weak peripheral COX inhibition (due to peripheral inactivation) → minimal anti-inflammatory activity
- Does not affect platelet function or bleeding time
Uses
- Fever and mild-moderate pain
- Preferred in: children with viral infections (no Reye risk), GI-sensitive patients, pregnancy (preferred analgesic), patients who don't need anti-inflammatory action
Metabolism and Toxicity
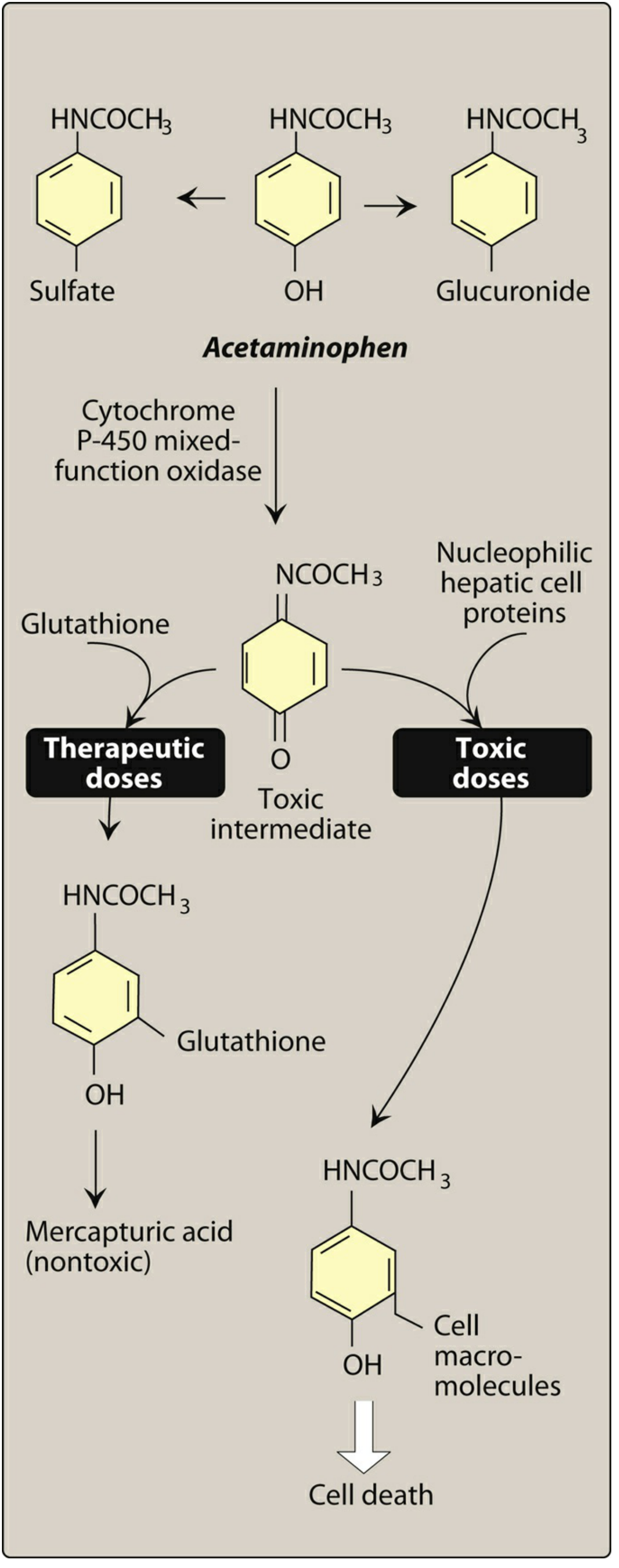
| Pathway | Normal Doses | Overdose |
|---|---|---|
| Primary | Conjugated to glucuronide and sulfate (inactive, excreted in urine) | These pathways become saturated |
| Minor (CYP450) | → NAPQI (toxic intermediate) → reacts with glutathione → mercapturic acid (nontoxic) | Glutathione depleted → NAPQI binds hepatocyte proteins → hepatic necrosis, cell death |
Toxicity Treatment
- N-acetylcysteine (NAC) - replenishes glutathione; most effective if given within 8-10 hours of overdose
- Acetaminophen overdose is a leading cause of acute liver failure in the developed world
Quick Comparison: NSAIDs vs Acetaminophen
| Feature | NSAIDs | Acetaminophen |
|---|---|---|
| Anti-inflammatory | ✓ | Minimal |
| Analgesic | ✓ | ✓ |
| Antipyretic | ✓ | ✓ |
| Antiplatelet | ✓ (aspirin strongest) | ✗ |
| GI side effects | ✓ (major concern) | Minimal |
| Renal effects | ✓ | Minimal |
| CV risk | ✓ (except aspirin) | Minimal |
| Hepatotoxicity | Rare | ✓ (overdose) |
| Safe in pregnancy | Avoid 3rd trimester | Preferred |
| Safe in children (viral infections) | ✗ (Reye risk with aspirin) | ✓ |
Source: Lippincott Illustrated Reviews: Pharmacology, 9781975170561
Gout
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
I now have the complete gout chapter. Here is the comprehensive, detailed answer:
Gout - Drugs for Treatment
Lippincott Illustrated Reviews: Pharmacology
I. Pathophysiology of Gout
Gout is a metabolic disorder characterized by elevated uric acid in the blood (hyperuricemia). The underlying cause is an imbalance between overproduction of uric acid and/or the inability to excrete uric acid renally.
Mechanism of Crystal-Induced Inflammation
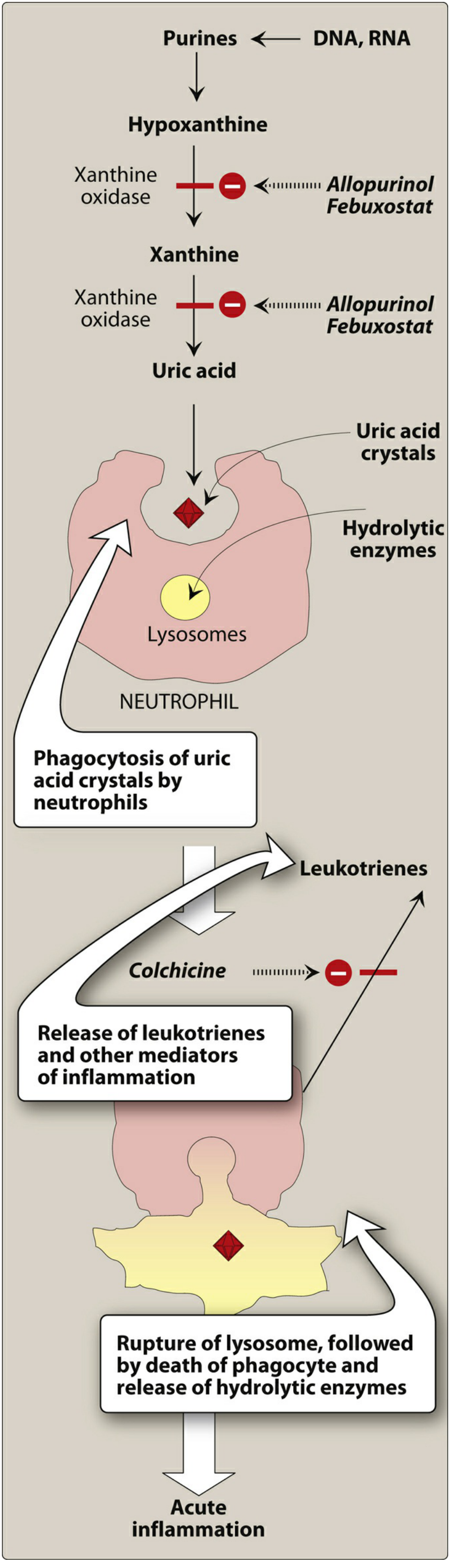
The cascade proceeds as follows:
- Purines (from DNA, RNA catabolism) → Hypoxanthine → Xanthine → Uric acid (via xanthine oxidase)
- Uric acid accumulates above the saturation point (>6 mg/dL) → deposits as sodium urate crystals in joints (especially the 1st MTP joint / big toe, knees, ankles, wrists, elbows) and kidneys (urate nephropathy, nephrolithiasis)
- Neutrophils phagocytize urate crystals → crystals resist digestion → lysosomal rupture → release of hydrolytic enzymes and leukotrienes → acute inflammation
- Colchicine interrupts this cycle by inhibiting neutrophil migration
Clinical Presentation
- Acute flares: sudden, severe pain, swelling, tenderness, warmth, and redness in the affected joint
- Triggered by: excessive alcohol, diet rich in purines (red meat, shellfish), dehydration, certain drugs (thiazide diuretics, low-dose aspirin), kidney disease, chemotherapy
Therapeutic Goal
Lower serum uric acid below 6 mg/dL (saturation point) to prevent crystal deposition. This is achieved by:
- Inhibiting uric acid synthesis (xanthine oxidase inhibitors)
- Increasing uric acid excretion (uricosuric agents)
II. Treatment Overview
| Phase | Goal | Agents |
|---|---|---|
| Acute gout attack | Reduce inflammation rapidly | NSAIDs, Corticosteroids, Colchicine |
| Chronic gout / prevention | Lower serum urate long-term | Allopurinol (1st line), Febuxostat, Probenecid, Pegloticase |
III. Treatment of Acute Gout
Acute attacks can be triggered by alcohol, high-purine diet, kidney disease, and more.
A. NSAIDs
- Indomethacin is the classic NSAID of choice for acute gout
- All NSAIDs are likely effective in decreasing pain and inflammation during acute attacks
- Use at maximum doses for shortest duration possible
- Avoid in patients with renal impairment (reduces renal blood flow), peptic ulcer disease, or those already on anticoagulants
- ⚠ Low-dose aspirin is contraindicated in gout - at low doses, aspirin decreases renal uric acid excretion and can worsen hyperuricemia
B. Corticosteroids
- Intra-articular corticosteroids are effective when only 1-2 joints are involved
- Systemic corticosteroids (oral prednisone) used when NSAIDs/colchicine are contraindicated or poorly tolerated
C. Colchicine (see full section below)
- Effective for acute attacks but NSAIDs have largely replaced it due to safety profile
- Must be given within 36 hours of attack onset to be effective
IV. Colchicine (Colcrys)
A plant alkaloid - not a uricosuric agent, not an analgesic; relieves pain specifically in gout through anti-inflammatory action.
Mechanism of Action
Colchicine binds to tubulin (a microtubular protein), causing its depolymerization. This:
- Disrupts neutrophil cytoskeleton → ↓ neutrophil motility and migration into inflamed joints
- Blocks phagocytosis of urate crystals by neutrophils → ↓ leukotriene and hydrolytic enzyme release
- Blocks cell division by binding to mitotic spindles (affects rapidly dividing cells)
The anti-inflammatory activity of colchicine is specific for gout - this selectivity has diagnostic value.
Therapeutic Uses
- Acute gout: usually relieves pain within 12 hours (must be given within 36 hours of onset)
- Prophylaxis against acute gout attacks during initiation of urate-lowering therapy
Pharmacokinetics
- Oral administration, rapidly absorbed from GI tract
- Metabolized by CYP3A4 in liver and other tissues
- Undergoes enterohepatic recirculation
- High interpatient variability in elimination half-life
- Portion excreted unchanged in urine
Adverse Effects
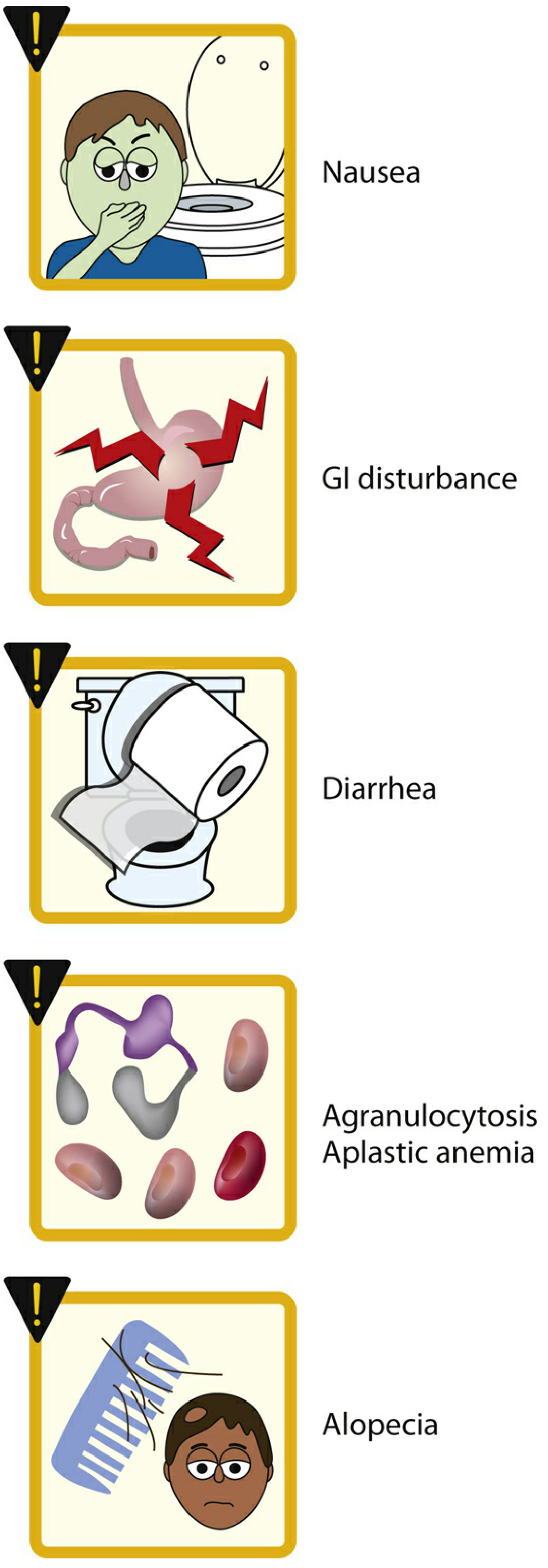
| Category | Effects |
|---|---|
| Acute (GI - most common) | Nausea, vomiting, abdominal pain, diarrhea |
| Chronic administration | Myopathy, neutropenia, aplastic anemia (agranulocytosis), alopecia |
| Contraindications | Pregnancy; caution in hepatic, renal, or cardiovascular disease |
Drug Interactions (Important)
- CYP3A4 inhibitors (clarithromycin, itraconazole) → ↑ colchicine levels → require dose adjustment
- P-glycoprotein (P-gp) inhibitors (amiodarone, verapamil) → ↑ colchicine levels → require dose adjustment
- Severe renal impairment → dose reduction required
V. Treatment of Chronic Gout (Urate-Lowering Therapy)
Key Principle:
⚠ Starting urate-lowering therapy can precipitate an acute gout attack due to rapid shifts in serum urate concentrations (crystal mobilization). Therefore, co-prescribe prophylactic low-dose colchicine, NSAIDs, or corticosteroids for at least 6 months when initiating any urate-lowering agent.
VI. Xanthine Oxidase Inhibitors - First-Line for Chronic Gout
These are first-line urate-lowering agents, blocking the last two steps of uric acid synthesis.
A. Allopurinol (Zyloprim) - Preferred first-line
Mechanism
A purine analog that competitively inhibits xanthine oxidase - the enzyme that converts:
- Hypoxanthine → Xanthine, AND
- Xanthine → Uric acid
Result: accumulation of hypoxanthine and xanthine (more soluble, more easily excreted) instead of uric acid.
Therapeutic Uses
- First-line urate-lowering therapy for gout (preferred over febuxostat and probenecid)
- Hyperuricemia secondary to malignancy (tumor lysis syndrome - especially post-chemotherapy when large amounts of purines are released)
- Hyperuricemia due to renal disease
Pharmacokinetics
- Completely absorbed orally
- Metabolized to alloxanthine (oxypurinol) - also an active xanthine oxidase inhibitor
- Alloxanthine has a half-life of 15-18 hours → allows once-daily dosing
- Drug and active metabolite excreted in urine
- ⚠ Dose reduction required if eGFR < 30 mL/min/1.73 m²
Adverse Effects
- Generally well tolerated
- Hypersensitivity reactions (skin rashes) - most common adverse reaction; risk increased in reduced renal function
- Allopurinol hypersensitivity syndrome (rare but severe): fever, exfoliative dermatitis (Stevens-Johnson syndrome/TEN), hepatitis, renal failure
- Acute gout flares more frequent during first months of therapy → use prophylactic colchicine/NSAIDs concurrently
Drug Interactions
- Azathioprine and 6-mercaptopurine - Allopurinol inhibits xanthine oxidase, which is responsible for their metabolism → markedly ↑ toxicity → dose reduction of azathioprine/6-MP by 50-75% required
B. Febuxostat (Uloric)
Mechanism
Non-purine xanthine oxidase inhibitor (structurally unrelated to allopurinol). Selectively inhibits both oxidized and reduced forms of xanthine oxidase.
Advantages over Allopurinol
- Less renal elimination → less dosage adjustment in renal impairment
- Lower risk of rash/hypersensitivity reactions compared to allopurinol
Disadvantages
- ⚠ Associated with greater risk of cardiovascular events (MI, stroke) compared to allopurinol
- Reserved for patients who have contraindications to, or cannot tolerate, allopurinol
VII. Uricosuric Agent
Probenecid (Generic only)
Mechanism
A weak organic acid that inhibits the urate-anion exchanger (URAT1) in the proximal tubule of the kidney. At therapeutic doses, it blocks tubular reabsorption of uric acid → ↑ urinary uric acid excretion → ↓ serum uric acid.
Use
- Second-line option for patients intolerant to xanthine oxidase inhibitors or with inadequate response
- Also used to increase serum levels of certain antibiotics (e.g., penicillins, cephalosporins) by blocking their renal tubular secretion
Limitations and Precautions
- ⚠ Avoid if creatinine clearance < 50 mL/min (inadequate urine flow to excrete the extra uric acid, risk of renal stones)
- ⚠ Low-dose aspirin antagonizes probenecid's uricosuric effect (aspirin at low doses promotes uric acid retention)
- Adequate hydration and urine alkalinization required to prevent urate stone formation
- Adverse effects: Nausea, vomiting, dermatologic reactions, rarely anemia or anaphylaxis
VIII. Recombinant Uricase
Pegloticase (Krystexxa) - Last Resort
Mechanism
Recombinant form of urate oxidase (uricase) - an enzyme present in most mammals but absent in humans. It converts:
Uric acid → Allantoin (water-soluble, non-toxic metabolite → excreted by kidneys)
This is a fundamentally different mechanism from inhibiting uric acid synthesis or increasing renal excretion - it directly degrades existing uric acid.
Indications
- Refractory/severe gout - patients who fail standard therapies (xanthine oxidase inhibitors)
- Can rapidly lower serum urate even in very high levels
Administration
- IV infusion every 2 weeks
- Premedicate with antihistamines + corticosteroids to reduce infusion reactions
Adverse Effects
- Infusion-related reactions (most important) - flushing, chest discomfort, dyspnea
- Anaphylaxis - risk highest in patients who develop anti-drug antibodies
- Gout flares may increase during early treatment
IX. Drug Summary: Mechanism Comparison
URIC ACID OVERPRODUCTION
└── Xanthine Oxidase Inhibitors
├── Allopurinol (1st line) - purine analog; competitively inhibits XO
└── Febuxostat - non-purine XO inhibitor; less renal adjustment needed
URIC ACID UNDEREXCRETION
└── Uricosuric Agent
└── Probenecid - blocks URAT1 in proximal tubule → ↑ renal excretion
DIRECT URIC ACID DEGRADATION
└── Recombinant uricase
└── Pegloticase - uric acid → allantoin (refractory gout)
ACUTE ATTACK MANAGEMENT
├── NSAIDs (Indomethacin - classic choice)
├── Corticosteroids (intra-articular or systemic)
└── Colchicine - binds tubulin → ↓ neutrophil migration
X. Key Drug Interactions Summary
| Drug | Interaction | Result |
|---|---|---|
| Allopurinol + Azathioprine/6-MP | ↓ XO-mediated metabolism of 6-MP | ↑ toxicity → reduce 6-MP dose by 50-75% |
| Allopurinol + Warfarin | ↓ Warfarin metabolism | ↑ INR → monitor closely |
| Low-dose Aspirin + Probenecid | Aspirin retains uric acid | Antagonizes uricosuric effect |
| Low-dose Aspirin in Gout | ↓ Uric acid excretion | Worsens hyperuricemia |
| Colchicine + CYP3A4 inhibitors | ↑ Colchicine levels | ↑ GI and bone marrow toxicity |
| Colchicine + P-gp inhibitors | ↑ Colchicine levels | Dose adjustment needed |
| Thiazide diuretics | ↓ Renal uric acid excretion | Precipitate/worsen gout |
XI. Clinical Pearls
- Serum uric acid target: < 6 mg/dL (some guidelines target < 5 mg/dL in tophaceous gout)
- Never start urate-lowering therapy during an acute attack - wait until attack resolves, then initiate with prophylactic colchicine cover
- Naproxen preferred if NSAIDs needed in gout patients with cardiovascular disease (least cardiovascular risk)
- Febuxostat preferred over allopurinol in renal impairment (less renal adjustment); but caution for CV risk
- Pegloticase is the most powerful urate-lowering agent but is reserved for refractory cases due to infusion reactions and cost
- Dietary advice: avoid high-purine foods (red meat, organ meats, shellfish), alcohol (especially beer), high-fructose corn syrup; maintain hydration
Source: Lippincott Illustrated Reviews: Pharmacology, 9781975170561
Rheumatoid arthritis
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Finding Sources
Reading File
Loading Image
I now have all the content needed for a thorough, complete answer. Here is the full RA chapter from Lippincott Pharmacology:
Rheumatoid Arthritis - Drug Treatment
Lippincott Illustrated Reviews: Pharmacology
I. Pathophysiology of RA
Rheumatoid arthritis (RA) is a chronic systemic autoimmune disease characterized by synovial inflammation leading to progressive joint destruction. The key drivers are:
- IL-1 and TNF-α - proinflammatory cytokines secreted by synovial macrophages
- These cytokines stimulate synovial cells to proliferate and synthesize collagenase
- Collagenase degrades articular cartilage, stimulates bone resorption, and inhibits proteoglycan synthesis
- Net result: pannus formation, erosion of bone and cartilage, joint deformity and loss of function
Diagnosis (Clinical Application 40.1)
| Feature | Detail |
|---|---|
| Joints involved | 3 or more joints; most common: PIP, MCP joints of hands, wrists, MTP joints of feet |
| Serology | Positive rheumatoid factor (RF) and/or anti-CCP antibodies |
| Inflammation markers | Elevated CRP or ESR |
| Duration | Symptoms present for >6 weeks |
| Exclude | Psoriatic arthritis, SLE, other arthritides |
Early diagnosis and early treatment are critical - the disease causes progressive joint destruction and loss of function if untreated.
II. Classification of Drugs Used in RA
| Class | Agents |
|---|---|
| Traditional DMARDs | Methotrexate (1st line), Hydroxychloroquine, Leflunomide, Sulfasalazine |
| Biologic DMARDs - TNF-α inhibitors | Adalimumab, Certolizumab, Etanercept, Golimumab, Infliximab |
| Biologic DMARDs - Non-TNF | Tocilizumab, Sarilumab (IL-6R antagonists), Abatacept (costimulation blocker), Rituximab (anti-CD20) |
| Targeted synthetic DMARDs (JAK inhibitors) | Tofacitinib, Baricitinib, Upadacitinib |
| Symptomatic agents | NSAIDs, Glucocorticoids |
Treatment Algorithm
DIAGNOSIS OF RA
↓
Start Traditional DMARD immediately
(Methotrexate preferred; alternatives: HCQ, Leflunomide, Sulfasalazine)
↓
Use NSAIDs / glucocorticoids as bridge therapy
↓
Inadequate response to methotrexate?
↓
Add Biologic DMARD (TNF-α inhibitor preferred) OR non-TNF biologic DMARD
OR add JAK inhibitor
↓
Combination: Methotrexate + Biologic DMARD most effective for remission
III. Traditional (Conventional) DMARDs
DMARDs (Disease-Modifying Anti-Rheumatic Drugs) slow disease progression, induce remission, and prevent structural joint destruction. They should be started as soon as possible after diagnosis.
A. Methotrexate (Otrexup, Trexall) - First-Line Preferred
Mechanism
A folic acid antagonist that:
- Inhibits cytokine production
- Inhibits purine nucleotide biosynthesis → leads to immunosuppressive and anti-inflammatory effects
Key Features
- Mainstay of RA treatment - the anchor drug for most RA regimens
- Response occurs within 3-6 weeks of starting treatment
- Dosed once weekly (unlike cancer dosing) → minimizes adverse effects
- Can be combined with traditional or biologic DMARDs when monotherapy is inadequate
Adverse Effects
| Effect | Detail |
|---|---|
| Mucosal ulceration | Most common (oral ulcers/stomatitis) |
| Nausea/GI upset | Common |
| Cytopenias | Particularly leukopenia |
| Hepatic | Cirrhosis with chronic use |
| Pulmonary | Acute pneumonia-like syndrome (methotrexate pneumonitis) |
✅ Folic acid supplementation reduces GI and hepatic adverse effects and improves tolerability - routinely co-prescribed. ⚠ Periodic monitoring: LFTs, CBC, signs of infection ⚠ Contraindicated in pregnancy (teratogenic, abortifacient)
B. Hydroxychloroquine (Plaquenil)
Mechanism
Unknown mechanism in autoimmune disorders (likely involves TLR signaling inhibition and lysosomal function).
Uses
- Early, mild RA - often used as initial therapy
- Frequently combined with methotrexate (triple therapy with sulfasalazine)
- Also used in SLE
Key Features
- Onset of effect: 6 weeks to 6 months (slow onset)
- Fewer adverse effects on liver and immune system than other DMARDs
Adverse Effects
| Effect | Notes |
|---|---|
| Ocular toxicity | Irreversible retinal damage (bull's-eye maculopathy) and corneal deposits - requires regular ophthalmologic screening |
| CNS disturbances | |
| GI upset | |
| Skin discoloration and eruptions |
C. Leflunomide (Arava)
Mechanism
An immunomodulatory agent that causes selective cell arrest of autoimmune lymphocytes. Its active metabolite (teriflunomide) reversibly inhibits dihydroorotate dehydrogenase (DHODH) - an enzyme necessary for de novo pyrimidine synthesis.
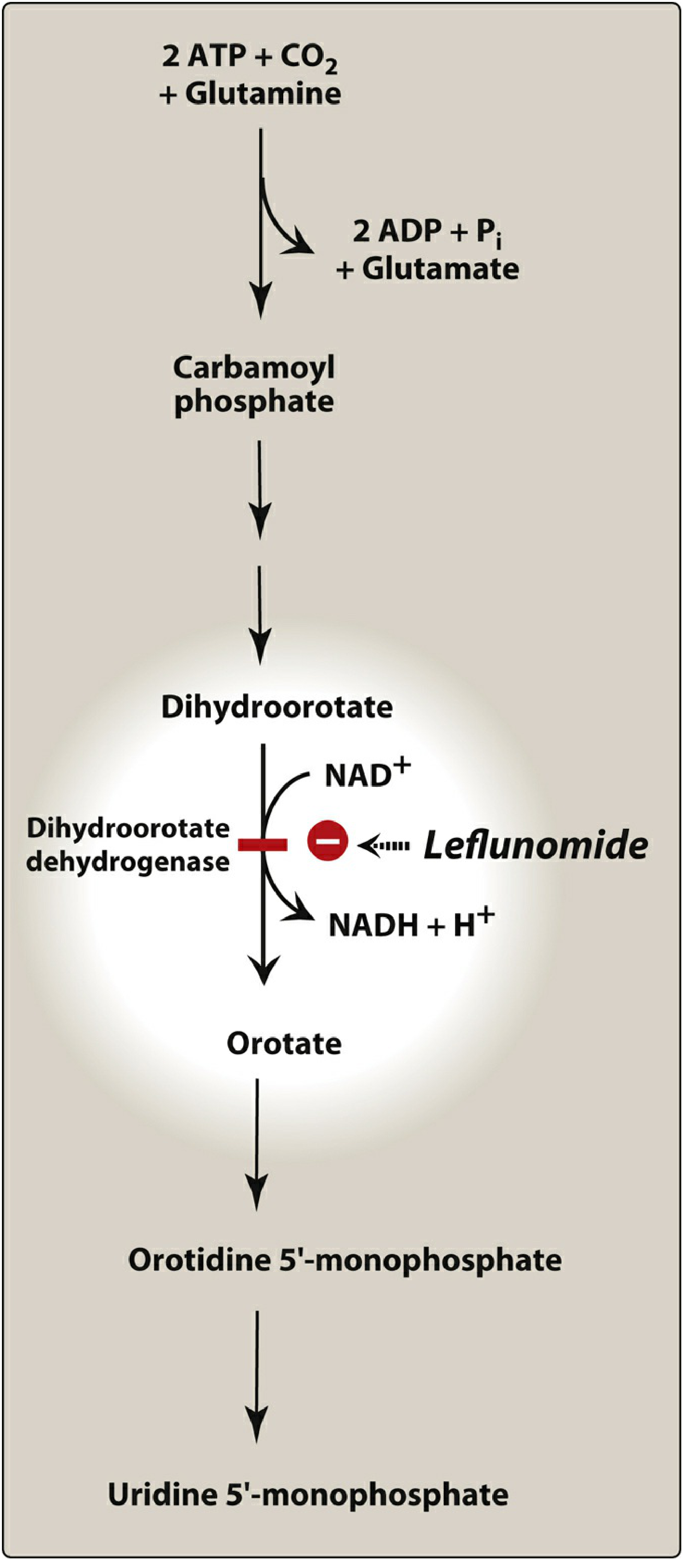
- Actively proliferating lymphocytes depend on de novo pyrimidine synthesis → leflunomide selectively arrests these cells
- Mature non-dividing cells can use salvage pathways and are less affected
Uses
- Monotherapy when methotrexate is not tolerated or contraindicated
- Combination with methotrexate for suboptimal methotrexate response
Adverse Effects
- Headache, diarrhea, nausea (common)
- Weight loss, flu-like syndrome, skin rash, alopecia, hypokalemia
- Hepatotoxicity - not recommended in liver disease; monitor liver enzymes
- Contraindicated in pregnancy
Monitoring
CBC, electrolytes, liver enzymes, signs of infection
D. Sulfasalazine (Azulfidine)
Mechanism
Unclear in RA (anti-inflammatory and immunomodulatory properties).
Key Features
- Onset of activity: 1-3 months
- Similar indications to leflunomide
- Used in mild-moderate RA; part of triple therapy with methotrexate + hydroxychloroquine
Adverse Effects
- GI (most common): nausea, vomiting, anorexia
- Leukopenia
E. Glucocorticoids (Symptomatic / Bridge Therapy)
- Potent anti-inflammatory (inhibit phospholipase A2, block arachidonic acid cascade, reduce cytokine production)
- Used to:
- Provide symptomatic relief during acute flares
- Bridge the gap while waiting for DMARDs to become effective (weeks to months)
- Always used at the lowest dose for the shortest duration possible to avoid long-term adverse effects (osteoporosis, adrenal suppression, hyperglycemia, Cushing syndrome, etc.)
- Intra-articular corticosteroids for 1-2 severely affected joints
IV. Biologic DMARDs
Biologics are large-molecule targeted therapies that specifically block inflammatory cytokines or immune cell activation. They are used when:
- Inadequate response to traditional DMARDs (especially methotrexate)
- Guidelines recommend adding a TNF-α inhibitor or non-TNF biologic DMARD in such cases
Class-Wide Safety Warnings (All Biologic DMARDs)
⚠ Increased risk of serious infections - TB, fungal opportunistic infections, sepsis ⚠ Screen for latent TB before starting any biologic (with TST/IGRA) ⚠ Hepatitis B reactivation risk - screen before initiation ⚠ No live vaccines during biologic therapy ⚠ Do NOT combine TNF-α inhibitors + non-TNF biologic DMARDs (severe infection risk)
A. TNF-α Inhibitors (5 agents)
Mechanism common to all: Block TNF-α → prevent binding to TNF-α receptors on cell surface → ↓ synovial inflammation, cartilage degradation, bone resorption
| Drug | Type | Route | Frequency | Notes |
|---|---|---|---|---|
| Adalimumab (Humira) | Fully human monoclonal Ab | SC | Weekly or every 2 weeks | Most widely used; broad indications |
| Certolizumab (Cimzia) | Humanized Ab Fab fragment (PEGylated) | SC | Every 2 weeks | PEGylation extends half-life |
| Etanercept (Enbrel) | Fusion protein (TNF receptor + IgG Fc) | SC | Once weekly | Generally well tolerated |
| Golimumab (Simponi) | Fully human monoclonal Ab | SC | Once monthly | Must combine with methotrexate |
| Infliximab (Remicade) | Chimeric (human/murine) monoclonal Ab | IV infusion | Every 8 weeks | MUST combine with methotrexate (prevents anti-drug antibody formation) |
TNF-α Inhibitor-Specific Adverse Effects
- Headache, nausea, rash, injection site reactions (SC agents)
- Infusion reactions with infliximab (fever, chills, pruritus, urticaria)
- Risk of lymphoma and other malignancies (Black Box Warning)
- Caution in heart failure (can cause/worsen HF)
- Reactivation of TB and hepatitis B
Combination Superiority:
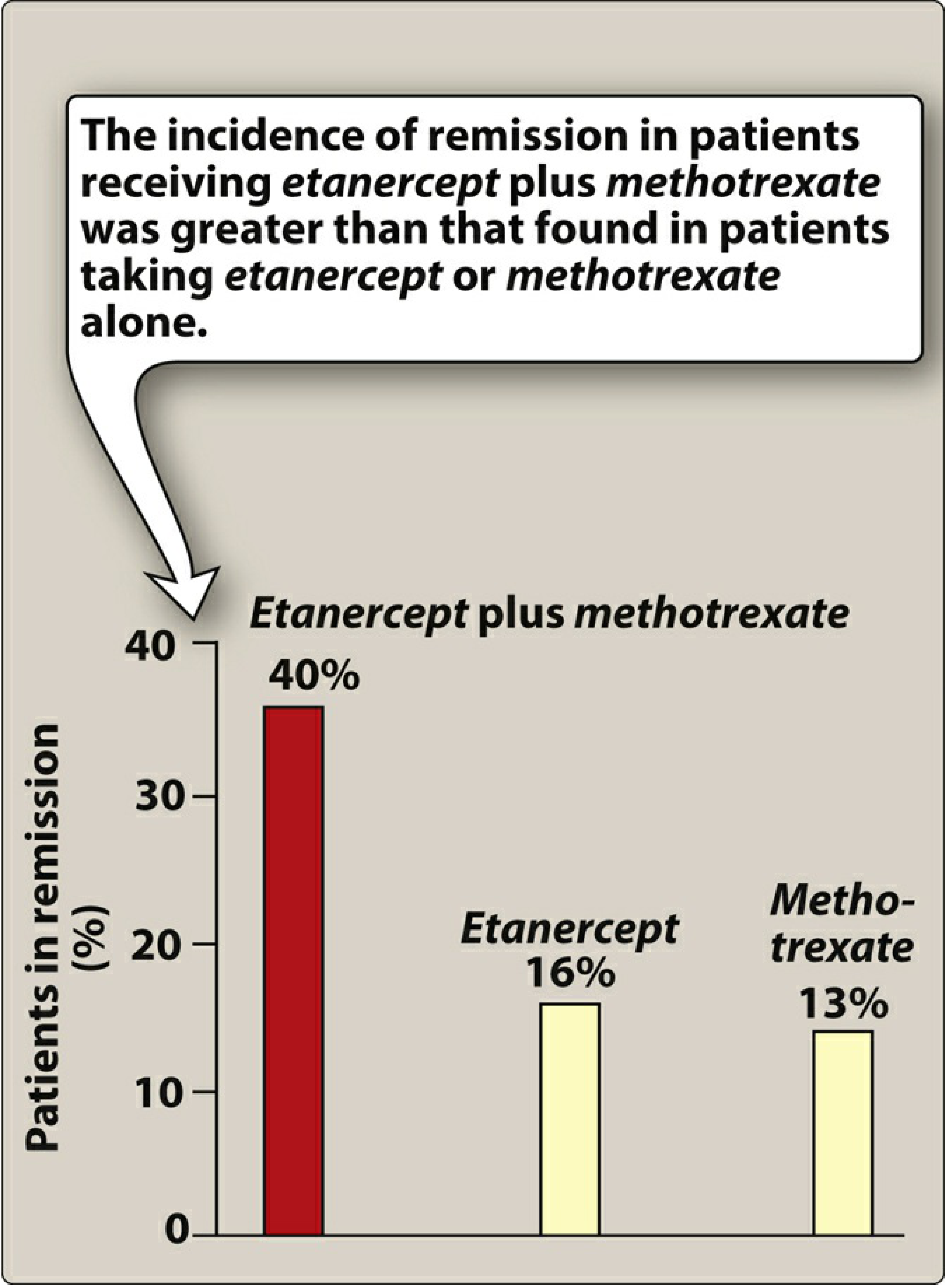
Etanercept + Methotrexate = 40% remission after 1 year vs etanercept alone (16%) and methotrexate alone (13%).
B. IL-6 Receptor Antagonists
Tocilizumab (Actemra) and Sarilumab (Kevzara)
- Recombinant monoclonal antibodies that bind to IL-6 receptors, inhibiting pro-inflammatory IL-6 activity
- IL-6 drives synovitis, acute-phase response (↑ CRP), and joint destruction
- Both can be used as monotherapy (unlike most other biologics which benefit from methotrexate combination)
- Tocilizumab: IV or SC; Sarilumab: SC
- Adverse effects: Neutropenia, thrombocytopenia, elevated LFTs, GI perforation (rare), increased infection risk
- Monitoring: CBC, LFTs, lipids
C. Costimulation Blocker - Abatacept (Orencia)
Mechanism
Abatacept is a fusion protein (CTLA4-Ig) that selectively modulates T-cell activation by blocking the CD80/CD86:CD28 co-stimulatory signal required for full T-cell activation. Without this second signal, T cells become anergic and cannot propagate the autoimmune response.
- Route: IV infusion monthly or SC weekly
- Response takes several weeks to months
- Preferred in patients with seropositive RA (RF+, anti-CCP+)
- Adverse effects: Headache, nausea, nasopharyngitis, increased infection risk
- Do NOT combine with TNF-α inhibitors
D. Anti-CD20 Antibody - Rituximab (Rituxan)
Mechanism
Rituximab is a chimeric monoclonal antibody that binds CD20 on B lymphocytes, leading to B-cell depletion (via complement-dependent cytotoxicity, ADCC, and apoptosis). B cells are important in RA as they produce rheumatoid factor and auto-antibodies, and act as antigen-presenting cells.
- Administered as IV infusion (two doses 2 weeks apart, repeated every 6 months)
- Used in RA refractory to TNF-α inhibitors or where TNF-α inhibitors are contraindicated
- Adverse effects:
- Infusion reactions (premedicate with corticosteroid + antihistamine + acetaminophen)
- Progressive multifocal leukoencephalopathy (PML) (rare, caused by JC virus reactivation)
- Hepatitis B reactivation
- Cytopenias, infection risk
V. Targeted Synthetic DMARDs - JAK Inhibitors
Janus kinases (JAK1, JAK2, JAK3, TYK2) are intracellular enzymes that mediate signaling from multiple cytokine receptors (including IL-6, IL-2, interferons, etc.) to the nucleus via the JAK-STAT pathway. JAK inhibitors block this intracellular signaling.
| Drug | Brand | Route |
|---|---|---|
| Tofacitinib | Xeljanz | Oral |
| Baricitinib | Olumiant | Oral |
| Upadacitinib | Rinvoq | Oral |
Indication
- Moderate-severe established RA with inadequate response or intolerance to methotrexate and TNF-α inhibitors
Pharmacokinetics
- All metabolized primarily by CYP3A4; dose adjustments needed with potent CYP3A4 inhibitors/inducers
Adverse Effects and Monitoring
| Effect | Details |
|---|---|
| Nausea, upper respiratory infections | Common |
| Anemia | Tofacitinib: Hb must be >9 g/dL to start; monitor during therapy. Baricitinib/Upadacitinib: avoid if anemia present |
| Lymphocytopenia / Neutropenia | Check lymphocyte and neutrophil counts before and during therapy |
| Malignancy | Increased risk of new primary malignancies (Black Box Warning) |
| Opportunistic infections | TB reactivation, fungal infections |
| GI perforation | Rare |
| Venous thromboembolism (VTE) | Increased risk (Black Box Warning, especially tofacitinib) |
Due to long-term safety concerns, JAK inhibitors are generally reserved for patients with inadequate response or intolerance to other agents.
VI. Other Agents Used in RA (Less Common)
Anakinra (IL-1 receptor antagonist), azathioprine, cyclosporine, gold salts, and minocycline are used infrequently due to:
- Adverse effect profile
- Availability of more effective/safer agents
VII. NSAIDs in RA
- Provide symptomatic relief (pain, stiffness, swelling) but do NOT modify disease progression
- Do not prevent joint destruction or induce remission
- Used adjunctively alongside DMARDs
VIII. Summary Comparison of All RA Drug Classes
RHEUMATOID ARTHRITIS TREATMENT
│
┌─────────────────────┼─────────────────────┐
│ │ │
SYMPTOMATIC TRADITIONAL BIOLOGIC/TARGETED
(No disease DMARDs (1st line) DMARDs (2nd line)
modification) │ │
│ │ ┌────────┴────────┐
NSAIDs Methotrexate TNF-α Non-TNF
Glucocorti- (preferred) Inhibitors Biologics
coids HCQ Adalimumab IL-6R: Tocilizumab
(bridge) Leflunomide Certolizumab Costimulation:
Sulfasalazine Etanercept Abatacept
Golimumab B-cell: Rituximab
Infliximab
JAK Inhibitors
(3rd line)
Tofacitinib
Baricitinib
Upadacitinib
IX. Key Drug Monitoring Requirements
| Drug | Monitor |
|---|---|
| Methotrexate | LFTs, CBC, renal function, signs of infection; supplement folic acid |
| Hydroxychloroquine | Ophthalmologic exam (retinal toxicity) every 1 year |
| Leflunomide | LFTs, CBC, electrolytes, signs of infection |
| All TNF-α inhibitors | Screen for latent TB (IGRA/TST), Hepatitis B serology before starting |
| Tocilizumab/Sarilumab | CBC (neutropenia), LFTs, lipid panel |
| JAK inhibitors | Hb >9 g/dL (tofacitinib), lymphocytes + neutrophils, signs of infection, VTE risk |
X. Contraindications Summary
| Drug | Key Contraindication |
|---|---|
| Methotrexate | Pregnancy, severe hepatic/renal disease |
| Leflunomide | Pregnancy, liver disease |
| Hydroxychloroquine | Pre-existing retinal disease |
| All biologics | Active serious infection, live vaccines, TB (until treated) |
| TNF-α inhibitors | Heart failure (caution), lymphoma history |
| JAK inhibitors | Anemia (Hb <9), active serious infection, high VTE risk |
Source: Lippincott Illustrated Reviews: Pharmacology, 9781975170561
This is a shared conversation. Sign in to Orris to start your own chat.